Craiguniversity.tk


This article appeared in a journal published by Elsevier. The attached
copy is furnished to the author for internal non-commercial research
and education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling or
licensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of the
article (e.g. in Word or Tex form) to their personal website or
institutional repository. Authors requiring further information
regarding Elsevier's archiving and manuscript policies are
encouraged to visit:


Author's personal copy
Journal of Chromatography A, 1218 (2011) 556–586
Contents lists available at ScienceDirect
Journal of Chromatography A
Chromatographic selectivity triangles
Andrew R. Johnson, Mark F. Vitha ∗
Department of Chemistry, Drake University, 2507 University Ave, Des Moines, IA 50311, USA
2010 marked the 50th anniversary of the use of selectivity triangles to characterize chromatographic
Available online 9 November 2010
phases. Such plots ultimately identify and quantify the blend of intermolecular interactions that occurbetween solutes and solvents/phases. The first chromatographic triangle was proposed by Brown and
applied to GC stationary phases. Snyder then developed the influential solvent selectivity triangle (SST)
Solvent selectivity triangle
based on the gas–liquid partition data of Rohrschneider. The SST was combined with simplex experi-
Micellar electrokinetic capillary
mental designs to optimize RPLC separations. Subsequent criticisms of the work revolved around the
inaccurate predictions that resulted from the SST. These inaccuracies ultimately relate to the inability
of the SST to account for the effects of water on the interaction ability of organic solvents. Other crit-
System selectivity cubeReversed phase liquid chromatography
icisms focused on the selection of the three probe solutes (ethanol, dioxane, and nitromethane) that
Gas chromatography
were used to define the apices of the SST. Here, the concerns include the lack of explicit consideration
of dispersion interactions and the fact that the three probes do not represent any single intermolecu-
Linear solvation energy relationship
lar interaction but rather reflect a blend of intermolecular interactions. The SST approach was modified
for NPLC by redefining the triangle apices to reflect the localization, general adsorption, and basicity ofNPLC mobile phase modifiers. Because water is generally absent in NPLC, the triangle approach leads tobetter predictions for NPLC than for RPLC. In subsequent modifications of selectivity triangles, Fu andKhaledi have created a micellar selectivity triangle (MST) based on linear solvation energy relationships(LSERs) and Zhang and Carr have used the Dolan–Snyder hydrophobic subtraction model to create RPLCcolumn selectivity triangles. We end this review by highlighting more recent methods for comparingselectivities and by discussing a new 3D visualization tool for classifying chromatographic systems ashaving similar or different fundamental energetics of retention and hence having similar or differentselectivities.
2010 Elsevier B.V. All rights reserved.
∗ Corresponding author. Tel.: +1 515 271 2596; fax: +1 515 271 1928.
E-mail address: [email protected] (M.F. Vitha).
0021-9673/$ – see front matter 2010 Elsevier B.V. All rights reserved.
doi:10.1016/j.chroma.2010.09.046
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Re-evaluation of the SST using solvatochromism and linear solvation energy relationships (LSERs) . . . . . . . . . . . . . . . . . . . . . . . 569
RPLC column selectivity triangle based on the hydrophobic subtraction model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 575
But they didn't fillthe desert with pyramids.
They just built some. Some.
They're not still out there,building them now.
The triangle has been used for tens of thousands of years
to represent many rich and complex ideas. Triangles sitting on
their base have represented the sun, maleness, and fire while
downwards pointing triangles have symbolized the moon, femi-
diabolize time. Right?
ninity, and water [1]. Alchemists used a horizontal line through
We must not curse the passage of time.
an upward triangle to symbolize air, and one through a down-
In this, Hecht suggests that while the form of the pyramid had
ward triangle to symbolize earth, thus creating triangular symbols
great symbolic and structural value, a time eventually came to seek
for the four elements: fire, earth, air, and water [2]. There-
new activities and new alternatives. Similarly, the chromatographic
fore, chromatography could be represented with these symbols
triangles that have been built have advanced our understanding
because water flowing through layers of earth can cause chemical
of selectivity and guided our selections of mobile and stationary
phases. In this review, we hope to shine light on those advances.
While the symbolism of triangles has a long history,
But we also illustrate the limitations of the technique and propose
the application of triangles to chromatography goes back a
a new alternative.
mere 50 years. This is quite short in absolute terms, but itrepresents half the life of chromatography [3]. Chromatogra-
2. The importance of the separation factor
phers adopted triangles, prisms, and pyramids for explanatorypurposes principally because they allow three or more col-
The separation factor, ˛ (formerly know as the selectivity factor)
umn characteristics to be incorporated in two-dimensional
is defined as kB/kA where k is the retention factor and A and B refer
to two solutes for which B elutes after A. The general resolution
We begin this review with a brief description of the importance
equation, which relates the plate count (N), the separation factor,
of selectivity in chromatography because many triangle schemes
retention factors, and resolution (R), shows that resolution is highly
aim at understanding the selectivity of one phase relative to oth-
dependent on the retention factor, particular at low ˛'s.
ers. The first report of triangles in chromatography is then discussed
to set the stage for all subsequent developments. We then exam-
ine Snyder's key solvent selectivity triangle and how it has been
adapted in various ways for the various modes of chromatogra-
For example, a change in ˛ from 1.1 to 1.2 nearly doubles the
phy (RPLC, NPLC, GC, MEKC). We end by departing from triangles
resolution, whereas it is necessary to increase the plate count
and propose a different geometric figure, the cube, for examining
four-fold for the same improvement in resolution. Thus, changes
and comparing selectivity. This shift can perhaps be best under-
to a chromatographic system that differentially affect the reten-
stood using an excerpt from Jennifer Michael Hecht's poem "On
tion of a critical pair of solutes are the key focus for improving
the Strength of All Conviction and the Stamina of Love" (from the
separations. For purposes of this review we are taking ‘system'
Next Ancient World published by Tupelo Press, copyright 2001.
to include the common variables such as temperature, station-
Jennifer Michael Hecht. Used with permission) [4] in which she
ary phase, and mobile phase composition that chromatographers
frequently change in order to affect selectivity.
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
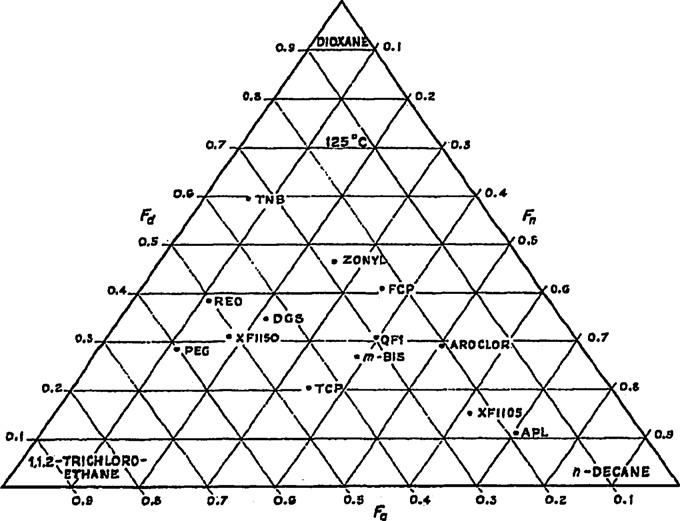
Fig. 1. Classification of GC stationary phases by Brown using dioxane, 1,1,2-
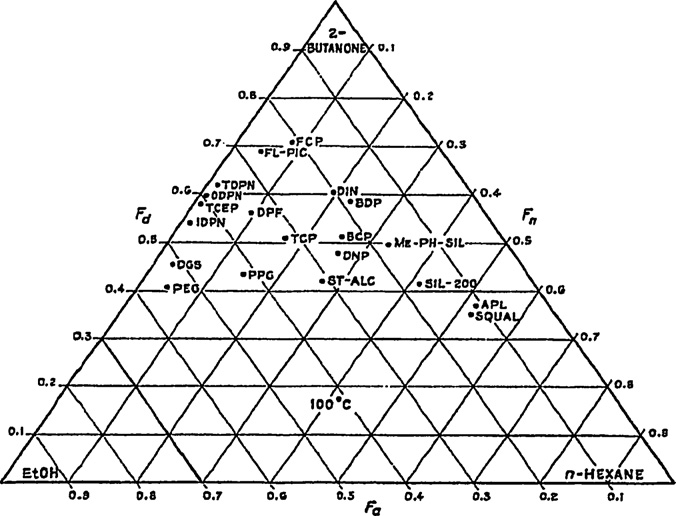
Fig. 2. Classification of GC stationary phases by Brown using 2-butanone, n-
trichloroethane, and n-decane as test solutes. F
hexane, and ethanol as test solutes. F
Fa, and Fd are retention
n , Fa , and Fd are retention fractions measured
fractions measured at 125 ◦C. APL = Apiezon L (Metropolitan, Vickers); XF 1105, XF
at 100 ◦C and 67 ◦C from literature sources available at that time. APL = Apiezon
1150 = cyanoethylated silicones (General Electric) with 5% and 50% cyano groups;
SQUAL = squalane;
Sil-200 = Silicone
Me-Ph-Sil = methylphenyl
AROCLOR = Aroclor 1262 (Monsanto) chlorinated biphenol; QF1 = fluorinated silicon
(Applied Science Labs); m-Bis = m-bis-(m-phenoxyphenoxyl)-benzene (Eastman);
BDP = benzyldiphenyl;
TCP = tricresyl phosphate (Albright and Wilson); Zonly = Zonyl E7 (Du Pont)
DIN = di-n-octyl ester of 4,4-dinitrodiphenic acid; TCP = tricresyl phosphate;
pyromellitic perfluoro ester; DGS = diethylene glycol succinate (Research Special-
PEG = polyethylene
PPG = polypropylene
ties Co.); PEG = polyethylene glyocol 1500 (Carbide and Carbon); REO = Reoplex 400
IDPN = imino-dipropionitrile;
(Geigy); TNB = 1,3,5-trinitrobenzene; FCP = diester of tetrachlorophthalic acid and
ODPN = oxydipropionitrile;
TDPN = thiodipropionitrile;
TCEP = 1,2,3-tris-(2-
cyanoethyl)-propane; FL-PIC = fluorine picrate; FCP = diester of tetrachlorophthalic
Reprinted from [6], with permission from Elsevier.
acid and 1-H,1-H,5-H-octafluoro-1-pentanol.
Reprinted from [6], with permission from Elsevier.
Comparisons of system selectivity try to help answer the ques-
tion: when a given system fails to achieve a desired separation,what does the analyst try next? Because all chromatographic
Brown also used different probe solutes. The results of creat-
separations are ultimately based on a blend of intermolecular inter-
ing the triangle based on n-hexane, ethanol, and 2-butanone are
actions (e.g., dipole–dipole, hydrogen bonding, and dispersion),
shown in Fig. 2. He noted that "the position of the triangular graph
using a system with similar blends of interactions as those demon-
for a given phase is determined by the choice of the three test
strated by the system that failed is unlikely to provide the desired
compounds, and these can be varied to suit a particular problem."
results. Instead, systems that are substantially different in their
The influence of the choice of probe solutes is important and will
intermolecular interactions must be sought. Thus, the questions
be raised elsewhere in this review with regards to characterizing
of 1) how to characterize systems in terms of their interaction abil-
LC-related systems.
ities and 2) how to differentiate one system from another naturally
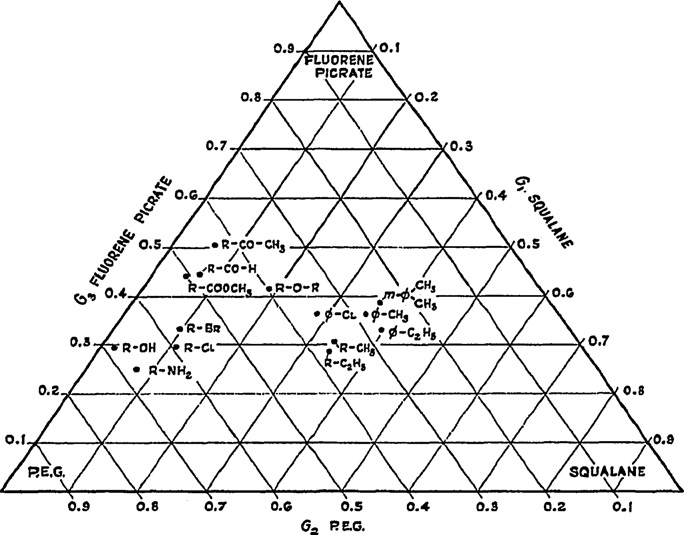
Interestingly, Brown then used an ‘inverse triangle' (current
arise. Selectivity triangle schemes that classify, differentiate, and
authors' description) to characterize the intermolecular interaction
group chromatographic systems have been used to help answer
abilities of individual steroids. This was done by selecting three
this question. In this review, we analyze various selectivity triangle
chemically different stationary phases – one neutral, one hydro-
schemes and how they have been applied to RPLC, NPLC, GC, and
gen bond (HB) accepting, and one HB donating – and using them to
MEKC systems.
define the apices of a triangle. The solutes were then characterizedby their affinity fraction, Ai, for each phase via the equation
3. The golden anniversary – 50 years of selectivity triangles
The year 2010 marked the 50th anniversary of the use of triangle
schemes to classify chromatographic systems. We make this state-ment based on the fact that the earliest report along these lines that
where ‘i' is one of the three columns represented by the numbers
we could find was from Brown in 1960 [5,6]. He created a triangle
1, 2, and 3. The three phases were SE-30 (silicone), NGS (neopentyl
to characterize GC stationary phases by defining a parameter, Fn, as
glycol succinate), and QF1 (fluorinated silicone).
Further, by taking the ratio of retention volumes of compounds
relative to retention values of an n-alkane of the same size, Brown
was able to make the plot shown in Fig. 3. The symbol G is used along
where Fn was called the ‘retention fraction', V represented reten-
the sides of the triangle because the ratio is ultimately related to
tion volumes, and i was n, a, or d which represented the retention
the free energy of retention of the functional group.
volumes of non-polar, electron accepting, and electron donating
Many of the ideas that Brown introduced would continue to
solutes. The solutes chosen to represent n, a, and d were n-decane,
appear in one form or another in subsequent papers using triangu-
1,1,2-trichloroethane, and dioxane, respectively. Each phase was
lar plots to characterize chromatographic systems. Interestingly,
thus characterized by three parameters that varied from 0.00 to
though, the exception to this is the application of the triangles in
1.00. The values were plotted at the apices of a triangle, resulting
an ‘inverse' manner for the purpose of characterizing individual
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Table 1
P� values for some common solvents [10].
Dimethyl sulfoxide
a Estimated values due to missing K values.
that dispersion interactions dominate gas–liquid partitioning, thatthis correction also accounts for dispersive forces that are not com-
Fig. 3. Identification of homologous series of compounds with various functional
pletely eliminated by the normalization of partition coefficients to
groups. G1, G2, and G3 group retention fractions measured at 100 ◦C and 67 ◦C
from literature sources available at that time. R = alkyl group; = phenyl group;PEG = polyethylene glycol.
Each of the solvents in Rohrschneider's collection was char-
Reprinted from [6], with permission from Elsevier.
acterized by a parameter, P�, defined as the sum of log K��g (P� =
and stronger hydrogen bond donating/accepting solvents generally
4. Snyder's solvent selectivity triangle
have higher P� values as shown in Table 1 [10].
The solvents were further characterized by normalizing log K��
4.1. General theory and development
for each test solute to P� according to
Brown's was the earliest report of triangle plots used to char-
acterize chromatographic systems, but it was Snyder's solvent
selectivity triangle (SST) published many years later that gen-
where i = e, d, or n for ethanol, dioxane, and nitromethane, respec-
erated more interest and critical examination [7]. Snyder based
tively, such that
his solvent characterization scheme on Rohrschneider's gas–liquid
partition coefficients for three test solutes – ethanol, dioxane, and
nitromethane – in 82 common solvents [8]. The three solutes werechosen to probe the ability of each solvent to participate in pro-
for all solvents.
ton acceptor, proton donor, and dipolar interactions, respectively.
Individual Xi values were used in a triangle plot to group the
However, as Cooper and Smith [9] point out,
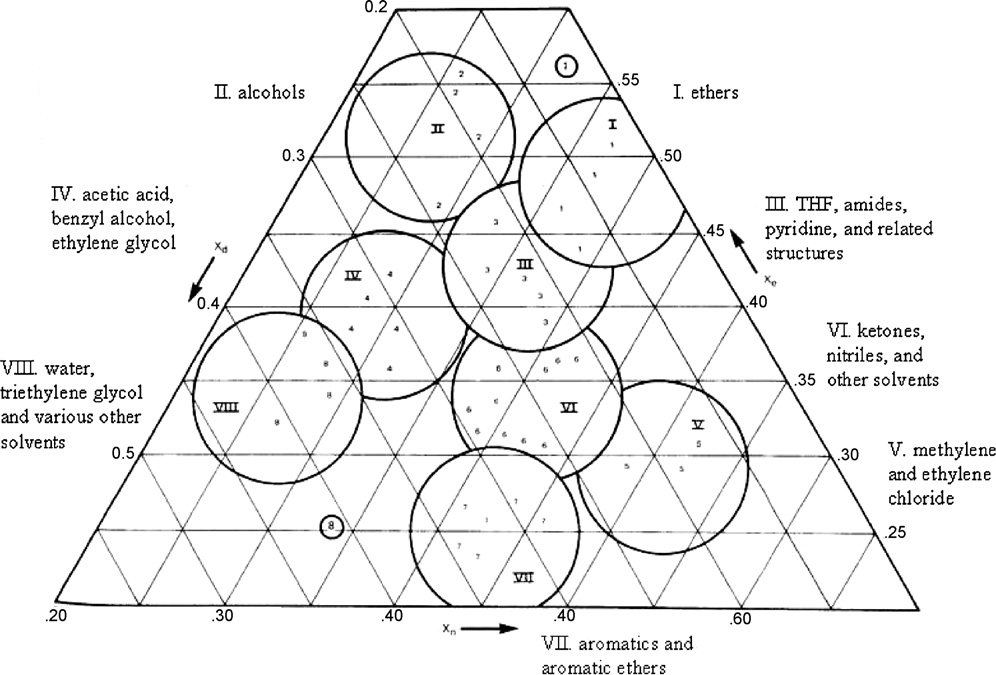
various solvents in Rohrschneider's data set. The resulting plot isshown in Fig. 4 [10]. It is worth noting that this plot is from a
". .in the Snyder system, ‘proton donor characteristics' actually
paper published in 1978, 4 years after the original publication,
refers to a solvent's ability to interact with a proton acceptor
because the actual X
(dioxane). It is not an actual measure of proton donating capa-
i values used to create the solvent triangle
in the original publication were inadvertently incorrect. In Fig. 4,
bility, and thus a solvent (or solute) can be classified as a proton
the circles represent groupings of common solvents. For example,
donor even though it contains no protons. The same qualifica-
group II is comprised of aliphatic alcohols (hence their relatively
tion applies to proton acceptors, which are classified as such
based on an ability to interact with a proton donor (ethanol)."
e values) and group VII is comprised of aromatic hydrocar-
bons, halo-substituted aromatic hydrocarbons, nitro compounds,
Put another way, the scales more broadly reflect Lewis acidity
and aromatic ethers – all highly polarizable compounds. The fact
and basicity rather than just interactions formally involving hydro-
that similar compounds fall close to one another in the triangle was
taken as evidence that the definition of Xi values does in fact reflect
To establish his characterizations, Snyder first corrected
actual chemical properties of the solvents and that the groupings
Rohrschneider's distribution coefficients for solvent molecular
are useful in identifying similar (or different) solvents in terms of
weight. These values were then normalized to the partition coef-
their ability to participate in specific intermolecular interactions.
ficient for a hypothetical alkane of the same volume in order to
Snyder's focus in the first publication was on solvents that could
remove the effects of dispersion interactions, which Snyder con-
be used in LC separations. The idea behind the triangle is that sol-
tends do not generally contribute significantly to selectivity. This is
vents in the same groups will provide comparable chromatographic
similar to Brown's taking the ratio of retention volumes for solutes
selectivity. Therefore, switching from one solvent to another within
to those of n-alkanes mentioned above. Snyder gave the resulting
the same group would not yield as dramatic a change in selectivity
values the symbol K��g. Lastly, a constant derived by considering the
as switching to a solvent in a group with very different characteris-
K��g values for each solute in saturated alkanes was subtracted from
tics (e.g., switching from group I to group VII or VIII). It is critical to
each value of log K��g to compensate for incomplete cancellation of
note that in this scheme, Snyder modified the traditional definition
dipole induced–dipole interactions, entropy, and other effects. A
of chromatographic selectivity with its focus on the separation of
different constant is used for each of the three test solutes. The
two different solutes in a particular solvent system, to one based
derivation of the constants is described in the original publications
on comparing two (or more) different solvents – or more broadly,
[7,10,11]. In essence, it makes the K��g values for each test solute very
two different chromatographic systems – and how they might sep-
close to zero in alkane solvents. It is reasonable to suggest, given
arate a set of solutes through different blends of intermolecular
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Fig. 4. Snyder's solvent selectivity triangle. Reproduced from the Journal of Chromatographic Science by permission of Preston Publications, a division of Preston Industries,
Inc. The solvent listings along the axes were added by the present authors.
interactions. For example, he differentiates between the strength
�Initromethane = 1.18�Initropropane, allowing for �Initropropane to be
of a solvent and its selectivity by stating "The strength of a solvent
calculated if it were not in the data sets of Rohrschneider [8] or
depends on its "polarity", or ability to preferentially dissolve more
McReynolds [12,13] for various phases. Based on these log K��g val-
polar compounds such as nitriles and alcohols. Solvent selectivity
ues, the parameters for Xi could be calculated for GC phases. Snyder
refers to the ability of a given solvent to selectively dissolve one
presented Xi values for diethylhexyl sebacate, diisodecyl phthalate,
compound as opposed to another, where the ‘polarities' of the two
tricresyl phosphates, carbowax 20, diethyleneglycol succinate, and
compounds are not obviously different" [7].
While the 1974 publication explained the derivation of the
Klee et al. [14] developed a selectivity triangle for GC phases
parameters and subsequent triangle plot, the 1978 publication is
(1) Most importantly, whereas the 1974 publication focused on
common, volatile organic solvents related to LC, the 1978 pub-
In an interesting modification of the SST for GC phases, they
lication was extended to include GC stationary phases,
used the sum of the three �Ii values to add another dimension to
(2) Snyder offers a defense of using just three test solutes to clas-
the triangle plot as shown in Fig. 5. This was done to indicate the
sify solvents. Two additional solutes (methylethyl ketone and
overall polarity of phases in addition to the relative importance of
toluene) were examined as part of this analysis,
the various specific interactions. Klee et al. also noted that for the
(3) Assertions are made regarding the relative unimportance of
best range of GC selectivities, it would be ideal to have phases with
dispersion interactions to selectivity, and
�I values in combination with points near the apices of the
(4) Snyder defends the groupings by showing the overall deviations
triangles, with the implication being that at that time, such a range
of Xi values from their averages are generally within 0.03 units
of phases was not available.
(one SD), or 0.015 if groups are further subdivided.
Thus, the 1978 publication simultaneously corrected, refined,
bolstered, and expanded the SST scheme presented in the 1974publication.
4.2. The SST and GC phases
As noted above, Snyder extended the SST to GC phases [10] by
using the conversion
where ‘i' is ethanol, dioxane, or nitropropane, �I
i = In,PH − In,SQ
where PH stands for the phase of interest and SQ repre-
Fig. 5. A selectivity prism in which the sum of retention indices (
nitromethane, and dioxane is used to add another dimension to a selectivity triangle
sents squalane, and ‘b' is the logarithm of the retention time
defined using those same solutes (see text for definitions of Xe, Xd, and Xn). CW-
increment per methylene unit added to a solute and is spe-
20M = Carbowax 20M.
cific to the phase being studied. It was further noted that
Reprinted from [14], with permission from Elsevier.
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
4.3. Teas diagrams
While the rest of this review focuses on the development, appli-
cation, and analysis of chromatographic selectivity triangles, webriefly note here that Teas [15] published a solvent triangle in theyears between the appearance of Brown's and Snyder's work. Histriangle was based on the work of Hansen [16] who used solubil-ity parameter and regular solution theory to define three solventparameters, ıd, ıp, and ıh to quantify the dispersion, polarity, andhydrogen bonding properties of solvents, respectively. Teas usedthese three parameters as the axes for his solvent triangle. Heapplied his triangle to make predictions about which solvents orsolvent mixtures would solubilize polymeric resins. It is interest-ing to note that Teas diagrams (as they are called) are used in thefield of art restoration to guide the selection of solvents to removevarnishes from old paintings [17]. For example, a Teas diagram wasused in the 1994 restoration of Johannes Vermeer's The Girl with aPearl Earring [18].
Fig. 6. Simplex experimental design involving seven training mobile phases (1–7)
and three mobile phases used to test the accuracy of the predictions. A, B, and C were
5. Impact of Snyder's solvent characterization scheme
mixtures of A = methanol/water (63:37%, v/v), B = tetrahydrofuran/water (39:61%,v/v), and C = acetonitrile/water (52:48%, v/v).
Reprinted from [19], with permission from Elsevier.
5.1. The chromatographic optimization factor
A number of publications using Snyder's solvent triangle as a
tion schemes involving three mobile phases, the three confirmatory
basis for optimizing chromatographic separations were published
analyses were dropped, leaving a seven-run optimization design. In
in the 1980s. The main impact of Snyder's work was in defining
the COF in this study, B = 0 and A = 1.0, indicating that time of anal-
three solvents that were deemed to have different selectivities. For
ysis was not a concern and the separation of all adjacent pairs was
example, in RPLC, methanol, acetonitrile, and tetrahydrofuran were
taken to be equally important. The COF was evaluated at three Rid
located in fairly distinct regions of the solvent triangle. For this and
values of 1.2, 1.8, and 2.4 and plotted in a triangle scheme shown in
other reasons, these solvents were used in addition to water to
Fig. 7. The optimum separation was found with 61%ACN : 39%THF.
optimize separations, often to the exclusion of other solvents.
Consistent with this, Glajch et al. proposed the chromatographic
5.2. Overlapping resolution mapping for RPLC
optimization factor (COF) as the basis for triangles related to max-imizing separations [19]. The COF is defined as
Due to limitations of the COF and difficulty extending it to
mixtures with more solutes, the authors developed overlapping
resolution maps (ORM) [19]. The ORM compares the resolution
of every pair of peaks in a chromatogram obtained for each sol-
vent mixture tested. A contour triangle map is used to estimate
the resolution for each pair in all compositions. Any area of the
i is the resolution of the ith pair of solutes in a mixture, Rid is
the ideal desired resolution, t
map with a resolution less than the desired resolution for that pair
M is the maximum acceptable analysis
is shaded in and areas with "excess" resolution are left clear. The
L is the experimental time. Ai is an arbitrary weighting
factor that allows greater emphasis on some critical pairs relative
maps for all adjacent pairs of compounds are overlaid and any area
to others. B is also an arbitrary weighting factor. The function is
that remains unshaded provides a solvent composition that could
constrained so that if R
separate the mixture with the desired resolution. Such an analysis
i > Rid then Ri is set equal to Rid, and if tM > tL,
tM is set equal to tL. Using these definitions and constraints, theCOF goes to zero for separations that meet all of the requirements.
Negative values indicate less desirable separations – the larger thenegative, the less desirable. This approach grew out of the chro-matographic response functions (CRF) of Morgan and Deming [20]and subsequent improvements proposed by Watson and Carr [21].
Glajch et al. acknowledge limitations of the COF as the basis forsolvent optimization. For example, it does not explicitly take notewhen peak elution order changes with different mobile phases. Fur-thermore, separations with overlapping peaks can have the sameCOF value as those with the expected number of peaks because themodel does not ‘know' how many peaks should be found – it simplymeasures the separation of the observed peaks.
Also in this report, a simplex design [22] involving ten test
runs, shown in Fig. 6, was used to optimize a three-solvent sys-tem (represented by A, B, and C) for a solute mixture of ninesubstituted napthalenes. In this figure, A, B, and C were mixturesof methanol/water (MEOH, 63:37%, v/v), tetrahydrofuran/water
Fig. 7. Chromatographic optimization factor (COF) plot based on simplex exper-
(THF, 39:61%, v/v), and acetonitrile/water (ACN, 52:48%, v/v),
imental design for the separation of nine substituted napthalenes. MEOH :methanol/water 63:37% (v/v); THF : tetrahydrofuran/water, 39:61% (v/v); and
respectively. Seven of the runs (labeled 1–7) were used to make pre-
ACN : acetonitrile/water, 52:48% (v/v). Conditions: 15 cm × 4.6 cm Zorbax-C8 col-
dictions of separations while the remaining three runs (8–10) were
umn, 2.0 mL/min, 40 ◦C, UV photometer, 254 nm.
used to test the accuracy of the predictions. In subsequent optimiza-
Reprinted from [19], with permission from Elsevier.
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Fig. 8. Overlapping resolution maps (ORM) based on simplex experimental design
for the separation of nine substituted napthalenes. Shaded areas represent sol-
vent mixtures that will not resolve the indicated pair to a resolution of 1.5.
The five most difficult solute pairs to resolve are shown in the plot. MEOH :
methanol/water 63:37%, (v/v); THF : tetrahydrofuran/water, 39:61% (v/v); and
ACN : acetonitrile/water, 52:48% (v/v). Conditions: 15 cm × 4.6 cm Zorbax-C8 col-
Fig. 9. A three-dimensional rendering of the ORM created by displaying the resolu-
umn, 2.0 mL/min, 40 ◦C, UV photometer, 254 nm.
tion for the most poorly resolved solute pair in the vertical direction. Based on the
Reprinted from [19], with permission from Elsevier.
data from the previous figure.
Reprinted with permission from [24]. Copyright 1983 American Chemical Society.
was performed with the retention data for the nine naphthalenederivatives to yield the triangle plot in Fig. 8 [19]. On this plot, the
competition for surface sites with solutes which can also localize.
optimum solvent mixture that was predicted by the COF method
The differences in the specific type of localization yield different
(designated with a ⊗) is included in the solvent mixture region
effects on selectivity. A third class of solvents which do not demon-
generated by ORM. The authors go on to demonstrate their method
strate localization effects, but rather appear to adsorb to the surface
using a literature data set of fifteen benzene derivatives [23].
in a more general manner was also identified. The three solvent
For this approach to work, it is necessary to perform peak match-
properties (i.e., non-localizing, localizing basic, and localizing non-
ing for each of the seven starting runs in order to identify any peak
basic) were used as the apices to create an NPLC-specific triangle.
cross-overs. Then the retention times and peak widths (or calcu-
Methylene chloride, MTBE, and ACN were used to represent the
lated peak widths) can be used to calculate the resolution of any
three properties, respectively, in a simplex optimization scheme.
critical solute pairs for every composition within the triangle.
We go into more detail about the influence of localization effects on
This approach was based on the optimization of only three
selectivity below. What is important to note here is the application
solvents (mixed with the fourth solvent, water). The choice of
of optimization schemes based on selectivity triangles to normal
optimizing three parameters was based on the conclusion from
phase separations.
the original SST work that only three general solvent charac-teristics affect selectivity. Better resolution may be achieved by
5.4. Gradient elution overlapping resolution mapping
including more solvents or optimizing any additional variablessuch as temperature that also influence selectivity. However,
Kirkland and Glajch extended the ORM approach to include gra-
including additional variables inflates the number of ‘training chro-
dient elution [26]. They did so by adding a third dimension – solvent
matograms' required by the simplex design, with a subsequent
strength – to the two-dimensional triangle plots. In the 2D plots, all
increase in the labor and time required to optimize the separation.
three apices were selected to have comparable solvent strengths.
In 1983, Glajch and Kirkland noted that the effects of different
Therefore, all that varied within the triangle space was selectivity.
stationary phases, temperature, pH, ionic effects, and secondary
By turning the triangle into a prism (see Fig. 10), solvent strength
equilibria such as ion-pairing could be incorporated into LC opti-
was added along the third dimension such that any vertical slice
mization schemes [24]. This publication includes a 3D visualization
of the prism parallel to the ends of the prism reflects mobile phase
involving triangle schemes (see Fig. 9). It resulted from adding the
systems of comparable solvent strength. Varying solvent strength
actual predicted resolutions in the third dimension rather than just
and selectivity allows gradient elution separations to be optimized
shading in regions below a certain threshold value as in the 2D
in much the same manner as described for isocratic optimizations.
triangle plots shown in the previous figure.
Seven mobile phase gradients were used to collect resolution datafor fourteen compounds. Estimates of resolution at other gradients
5.3. Overlapping resolution mapping for NPLC
were obtained via quadratic equations based on the original sevencompositions and used to create resolution contour maps for indi-
Glajch et al. extended the ORM approach to optimizing the
vidual pairs of solutes. An overlapping resolution map (now 3D)
NPLC separation of thirteen substituted naphthalenes on bare sil-
then indicates the position along the gradient and the solvent com-
ica particles [25]. The selection of the three mobile phase additives
position that yields the maximum predicted resolution. While each
(methylene chloride, acetonitrile, and methyl tert-butyl ether, all
slice of the prism represents a different solvent strength, proceed-
mixed in hexane) was based on a new triangle scheme designed to
ing through the prism along any one line of solvent strength (e.g.,
account for effects that are important in NPLC. Specifically, basic
line 7 in Fig. 11) does not change the selectivity of the mobile phase
polar solvents (e.g., methyl tert-butyl ether, MTBE) localize on
[26]. Thus, analyses using such gradients were termed ‘isoselec-
the solid surface through direct hydrogen bonding with the sur-
tive multisolvent gradient elution' (IMGE). The authors note that
face. Other solvents with diminished basicities, such as acetonitrile
the chromatogram obtained with the predicted gradient achieved
(ACN), also localize on the surface but in a different manner than
a resolution of 2.0 or greater for the fourteen compounds in under
do the basic polar solvents. Both types of solvent localization create
fifteen minutes (15 cm × 0.46 cm column, Zorbax C-8, 3.0 mL/min,
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Fig. 10. Solvent strength prism for gradient elution with an isocratic selectivity
triangle for one solvent strength. Apices are methanol (MeOH), acetonitrile (ACN),
and tetrahydrofuran (THF).
Reprinted from [26], with permission from Elsevier.
35 ◦C, particle size not shown) and that this was better than any ofthe seven gradients used to establish the analysis.
It is more typical in gradient elution to simultaneously vary
selectivity and solvent strength. Kirkland and Glajch used the term‘selective multisolvent gradient elution' (SMGE) to describe thisapproach [26]. Visual inspection of the seven initial chromatogramsresulted in a gradient depicted in Fig. 12. The chromatogramobtained with this gradient yielded even better separation for allpeaks and resulted in a different elution order for some of thepairs. The authors did note, however, abrupt baseline changes cor-responding to the abrupt changes in mobile phase compositiondepicted in the figure. Nonetheless, with seven training gradientsselected based upon Snyder's original selectivity triangle (to selectthe three organic solvents) and simplex experimental design pro-
Fig. 12. Representation of solvent program for step-selectivity gradient solvent
system. (—) Water; (· · ·) methanol; (- - -) acetonitrile; (-·-) tetrahydrofuran.
tocols, the authors were able to systematically select a quaternary
Reprinted from [26], with permission from Elsevier.
mobile phase gradient that allowed for complete separation of allcompounds.
presented an approach that slightly reduced the complexity of
5.5. Additional work on optimizations
calculation and the number of training chromatograms required(down to four) to obtain optimal solvent strength and selectivity for
Shortly after Kirkland and Glajch published the prism scheme
isocratic separations. They demonstrated this approach with a rela-
for optimizing gradients elution, Sticher and co-workers [27]
tively simple mixture of four flavonoid glycosides. Different groupshave suggested from four to ten or more training experiments.
The choice naturally depends on the accuracy of the predictionsthat is required. More training experiments will be required forgreater accuracy, separations with larger numbers of analytes, andseparations involving analytes with closely related structures.
Whereas Glajch, Kirkland, Squire, and Minor's ORM approach
strives to obtain a solvent mixture that maximizes COF (related toln[Ri/Rid]) for all components (if weighting factors are not used),O'Hare and co-workers modified this approach to focus on relativeretention rather than on absolute retention as a function of solventcomposition [28,29]. In their reports, the parameter that is relatedto solvent composition is ln(RTo/RTn) where RTo is the retentiontime of an internal standard and RTn is the retention time of various
Fig. 11. Experimental design for seven gradient elution chromatograms to obtain
components. Separate polynomial equations are obtained for each
data for optimization calculations. See original reference for solvent compositions.
compound in the mixture based on seven training chromatograms
Reprinted from [26], with permission from Elsevier.
selected in a manner akin to that used by Glajch et al. based on
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
SST to RPLC arise largely because of the effects of water, althoughchallenges were also made to the application of triangles to NPLCand GC. In RPLC, water is present in varying amounts in the mobilephase. This has three main effects: (1) increasing water contentincreases the overall polarity of the mobile phase and thereby altersthe selectivity of the separation, (2) the water modifies the abilityof the organic mobile phase additives to interact with solutes andthese alterations affect different solutes to different extents and (3)the water itself interacts differently (i.e., selectively) with differentsolutes. None of these effects is captured in the SST because the SSTwas based on pure organic solvents, not solvents modified withwater.
The work of Carr and co-workers [30,31] and El Seoud and co-
workers [32] illustrates some of these complexities. Non-linearitiesin the frequency of maximum absorbance of solvatochromic dyesvs. percent water in methanol/water and acetonitrile/water mix-tures are observed. These non-linearities are attributed to bothmicroheterogeneity and to preferential solvation effects [30–32].
Furthermore, the nature and extent of these effects depend bothon the organic modifier and the composition of the mixture. Forexample, acetonitrile/water mixtures were found to be dominatedby solvent clustering between 30% and 80% acetonitrile [32–37].
In methanol/water mixtures, however, Shulgin and Ruckenstein[38] assert that if any clusters exist, they are small. While solventclustering may not be extensive in methanol/water mixtures, the
Fig. 13. Statistical design for optimizing isocratic elution using four solvents. The
spectroscopic studies of Carr and co-workers [30,31] and El Seoud
numbers indicate individual mobile phases in which retention data for all solutes
and co-workers [32] suggest that preferential solvation of solutes
must be obtained. The authors note that in RPLC, some water-rich and water-
may still occur. Regardless of which effects exist within specific
poor phases can be eliminated, reducing the number of training chromatogramsto twelve.
aqueous mixtures, neither the effects of microheterogeneity nor
Reprinted from [28], with permission from Elsevier.
preferential solvation on solute retention are incorporated in theSST. Thus, the SST may not produce accurate predictions of selec-
simplex designs. The authors stated interest was in analyzing mix-
tivities when aqueous mobile phases are used. Examples of this are
tures of adrenocorticol steroids, with a primary requirement "to
discussed below.
separate and measure aldosterone without interference from other
The failures of the SST arising from the presence of water in
unrelated steroids [that were in the mixture] together with the
RPLC do not carry over to NPLC because the water content in NPLC
resolution of 18-hydroxysteroid congeners of aldosterone, 18OH-B
mobile phases is generally minimized. Thus, predictions of NPLC
and 18OH-A." This goal necessitated the shift from overall reso-
mobile phase selectivity based on triangle schemes, when specific
lution to one that required specific attention on critical solutes,
solvent localization and basicity effects are taken into account, are
hence the emphasis on individual retention times rather than on
generally much more reliable than those in RPLC.
resolution mapping for all components obtained via an ORM. The
The other major challenges considered in this section revolve
authors acknowledge that ORM can be adjusted to focus on crit-
around (1) the influence of interfacial adsorption and inadequate
ical analytes by excluding solvent selectivity areas corresponding
retention of the test solutes in GC and (2) the number and spe-
to pairs of minor importance, but they noted some problems asso-
cific nature of the test solutes used to create selectivity triangles.
ciated with this for their particular sample of interest. Using their
The focus here is on the importance of incorporating dispersion
approach, they were able to identify a mobile phase composition
interactions and the influence that using different test solutes has
that achieved their goals.
on the position of solvents within the triangle (i.e., their overall
Interestingly, they extend their analysis to consider the require-
classification and grouping).
ments of optimizing four-component systems (the above studieshave four-components – water, methanol, THF, and ACN – but each
6.1. Steroids and polystyrene oligomers
apex of the triangle upon which the approach is based is actuallya mixture such as water/MeOH, etc.). A four component system
While Snyder's solvent selectivity triangle had an important
could include the four pure solvents, or perhaps involve another
impact on LC solvent selection as demonstrated by the above opti-
water/solvent mixture such as water/dioxane. If a four-component
mization methods, others discussed the limitations and failures of
system were considered, simplex optimization dictates the need
the approach. For example, West described the failure of the solvent
for fifteen training chromatograms as shown in Fig. 13 [28]. The
selectivity triangle to group solvents according to their selectivity
time requirements and subsequent complexity of the data analy-
for resolving aromatic compounds and steroids using RPLC [39,40].
sis for such an optimization become much more cumbersome than
Lewis et al. made the same observation for polystyrene oligomers
those for ternary systems and often are unnecessary, particularly
because most of the theoretical optimizations we have focused on
In his work related to steroid separations in RPLC, West noted
here result in isocratic mobile phases and therefore do not take
that the slopes of steroid retention factor (measured using 2-
advantage of the practical benefits of gradient elution.
ketoalkanes as standards akin to Kovats GC-based retention indicesusing n-alkanes) vs. volume fraction of organic solvent showed con-
6. Failures of and modifications to the selectivity triangle
siderable variability for solvents from the same selectivity group.
Specifically, he noted the average slope for twelve steroids was 2.3
In this section we discuss challenges to the SST that appeared
times greater for 1-propanol than for methanol, which are in the
in the literature. The problems that were found when applying the
same solvent group in the triangle. He also noted that the slopes
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
were sometimes more similar for solvents in different groups than
contribute to experimentally observed selectivity for more com-
within groups. For example, the average slope (again over twelve
plex molecules." [40]
steroids) for 2-methoxyethylacetate (Group VI) was closer to that
In fairness to Snyder's selectivity triangle, it must be pointed out
for tetrahydrofuran (Group III) than it was for acetonitrile (also
that it was not intended to be used in the way West applied it. It
Group VI). Other such examples are provided in his article [40].
was a general scheme for classifying solvents to facilitate the selec-
The resolution of particular pairs of steroids in aqueous mobile
tion of solvents that are broadly different in the way they interact
phases with different organic modifiers of comparable solvent
with a wide range of solutes of varying chemical characteristics. It
strength was also studied. For spironolactone and ethisterone, the
was not designed to predict the best solvent for resolving individ-
resolution obtained with 2-ethyoxyethanol, 2-methoxyethanol,
ual pairs of closely related solutes. Nevertheless, West's findings
and tetrahydrofuran (all in Group III) was 0.68, 1.15, and 3.26,
call into question the overall similarity of some of the solvents in
respectively. It should be noted that the mobile phase com-
various groups, as well as highlight the potential effects of water
position was adjusted such that the first peak eluted with a
and dispersion interactions on selectivity (see below for more on
retention factor of 2.00 ± 0.03 to ensure comparable mobile phase
the topic of dispersion).
strengths. Within Group VI solvents, the resolution with diox-
The work of Martire and co-workers [37] is interesting as it
ane, 2-methoxyethylacetate, and acetonitrile was 1.70, 0.87, and
relates to West's criticism that the SST fails to account for the role
0.59, respectively. West also notes that the resolution obtained
of the stationary phase. Using alkylbenzenes as test solutes, activ-
with solvents from different groups is often more similar than
ity coefficients from the literature, and experimental measures of
that obtained with solvents within the same group. For example,
retention volumes, Martire et al. calculated contributions to the
the resolution of prednisone and hydrocortisone in THF (Rs = 1.81)
methylene unit selectivity arising from the mobile and station-
and 2-methoxyethanol (Rs = 1.93), both from Group III, was more
ary phases as a function of percent modifier in methanol/water
similar in ethanol (Rs = 1.88) from Group II than in another group
and acetonitrile/water mobile phases. They show that the sta-
III solvent, 2-ethoxyethanol (Rs = 2.54). Similar observations were
tionary phase contribution with both modifiers is comparable in
made for spironolactone and ethisterone.
magnitude and essentially constant from 5% to 60% water. The con-
West states that these observations "contradict the theory of the
tribution from the mobile phase, however, varies significantly over
solvent selectivity triangle concept" [40] and then goes on to sug-
that range, and is considerably larger than the stationary phase
gest that the discrepancies result from the fundamental assumption
contribution at all compositions. Tan and Carr provide a compa-
that dispersion interactions do not play an important role in deter-
rable result based on the analysis of mobile and stationary phase
mining solvent selectivity for solutions of polar solvents. Certainly,
cohesive energy densities for systems involving methanol, ace-
given the structural similarity of the steroids in this study, it is
tonitrile, and tetrahydrofuran. They state that "As the fraction of
reasonable to suggest that their overall characteristics regarding
water is increased, the cohesive energy density of the mobile phase
polarity and hydrogen bonding are comparable enough that even
increases substantially. However, changes in the cohesivity of the
small differences in dispersion interactions in the solvents, if not
bonded phase, which are largely controlled by the sorbed solvent,
accurately corrected for, could play a critical role in solvent selec-
are minor" [42]. These results suggest that assuming a constant (and
relatively unimportant) contribution to solvent selectivity arising
Focusing on dispersion only, however, neglects the more impor-
from different modifications of the stationary phase due to differ-
tant effects that water has on solvent selectivity. Specifically,
ent organic additives may be a reasonable approximation. Here
Snyder's groupings are based on Rohrschneider's data, which were
again, it is important to remember that the SST ultimately deals
collected for pure solvents. In contrast, West used binary mix-
with solvent selectivity. Thus, while the stationary phase clearly
tures of solvents with water as the diluent. It is well known that
makes an important contribution to the overall retention of solutes,
water is hardly an ‘inert' solvent and can significantly alter the
stationary phases modified with different solvents may be compa-
properties of bulk organic solvents. Furthermore, it does so in dif-
rable enough in their characteristics that differences in the mobile
ferent ways depending on the organic solvent and the percent
phases alone are more important to overall selectivity differences.
composition of the mixture as discussed above with regards to pref-
If this is the case, West's concerns about the role of the stationary
erential solvation and microheterogeneity. These variations could
phase may be overstated. We note here, however, that the changing
very well cause a difference between the group that a pure sol-
structure of the stationary phase and the modification of the alkyl
vent would be in compared to that of aqueous mixture of the
chains and surface silanol groups by sorbed solvents is clearly an
same solvent. The adjustment of solvent strength to obtain a reten-
important aspect of RPLC retention. Tan and Carr [42] provide an
tion factor of 2.00 for the earliest eluting peak is an arbitrary
extensive discussion of the influence of sorbed water and modifier
choice and required different amounts of water for different sol-
on mobile and stationary phase properties and how they contribute
vents. Clearly, the amount of water and its alterations of organic
to changes in solute retention. An analysis of the effects of solvent
solvent characteristics will significantly impact retention of polar
sorption in general, and of their work in particular, is outside the
and hydrogen bonding solutes as compared to its impact on non-
scope of this review, but the reader is encouraged to consult their
polar compounds. Thus, varying amounts of water will influence
the selectivity of the separation in ways that the SST scheme
In fairness to West, it must be noted that he acknowledged
for pure solvents does not incorporate and cannot accurately
the possibility that the structural similarity of the steroids and
polystyrene oligomers used in previous studies was the major fac-
West does not comment directly on the influence that different
tor behind the discrepancies between groupings and selectivities
amounts of water in the mobile phase have on selectivity. But in
that he observed. To address this, he conducted another study with
recognition of the possibility that dispersion plays an important
sixteen aromatic compounds (13 monosubstituted and three posi-
role in selectivity, and also in consideration of the assumption that
tional isomers) using aqueous mobile phases of twelve solvents
the stationary phase does not affect separations, he states
ranging in P� values from 3.9 to 7.2 from three groups in the solvent
"Perhaps these assumptions have resulted in an oversimplified
triangle (II, III, and VI). The binary mobile phases were adjusted to
approach to characterizing selectivity, or perhaps the three test
yield retention factors of 4.00 ± 0.04 for benzene in an effort to keep
solutes that were used to establish the solvent triangle do not
solvent strength constant. Again West used retention indices based
adequately encompass all of the important characteristics that
on 2-ketoalkanes to measure retention. He noted that the retention
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
indices of some compounds in some solvents were more compa-
the molar volume and the dispersion-related solubility parameter
rable in solvents from different groups than in solvents within the
for the solutes, and (4) differences in the solvents' solubility param-
same group, leading him to state "in general, there was very lit-
eters. In others words, according to this theory, dispersion effects
tle or no correlation between retention indices and the solvents
do not cancel as they relate to solvent selectivity. The extent of
grouped according to the selectivity triangle concept" [39]. He also
their importance depends on the combination of solutes and sol-
measured resolution of various compound pairs, noting
vents being considered. We did some simple calculations involvingvarious combinations of hexane, pentane, benzene, and toluene as
"the results of this study confirmed that solvents in the
solutes and solvents. The most dramatic effect was observed using
same selectivity group seldom give similar resolution, even at
hexane and toluene as solutes and pentane and benzene as solvents.
consistent solvent strength. .Numerous examples of extreme
In this case, our calculations using regular solution theory sug-
variation of R with the solvent groups are evident, with res-
gest that the selectivity for these solutes in pentane will be nearly
olution frequently being more alike for solvents classified in
four times greater than in benzene. Using benzene and toluene as
different groups than for those within a given group."
solutes and hexane and pentane as solvents led to the result that
Here again, different amounts of water were required to achieve
the selectivity in pentane will be only 1.0026 times greater than the
comparable solvent strengths for the elution of benzene. As noted
selectivity in hexane. So even from these systems, in which disper-
above, water preferentially alters the selectivity of polar and hydro-
sion is the only dominant intermolecular interaction, it is difficult
gen bonding solutes. It does so through the three mechanisms
to state how important dispersion interactions are to determining
discussed in the introduction to this section, namely, a general
solvent selectivity. It can be said that they do not cancel, but the
increase in mobile phase polarity with increasing water, modifi-
magnitude of their effect varies with specific systems.
cation of the solvent interaction abilities, and direct interaction
Two things must be noted. First, we have considered systems
with solutes. This suggests that an expansion of Snyder's triangle
in which dispersion is the main intermolecular interaction. It may
to include mixed solvents would provide valuable chemical insight
be that the contributions of dispersion to solvent selectivity are
into the effects of water on the properties of common organic sol-
quite small compared to the contributions from dipole–dipole and
vents. It would also increase the predictive power of the triangle
hydrogen bond interactions when polar and hydrogen bonding
with practical implications for RPLC.
solutes and solvents are considered. Second, the above results
West, however, might reject this idea as his writings indicate a
were based solely on regular solution theory with no further nor-
fundamental objection to the construction of the triangle, namely
malization or attempts to cancel dispersion interactions. Snyder,
that "it is constructed using data that does not correlate with
however, in the development of the triangle, corrected Rohrschnei-
resolution" and that specifically "the use of fractions of summed
der's partition data for differences in solvent molecular weight
retentions actually serves to hide differences in selectivity by mask-
and then normalized the results to the partition coefficient for a
ing absolute differences in retention units" [39]. He notes that these
hypothetical alkane of the same volume. Following this, a con-
criticisms extend to the classification of NPLC solvents and GC sta-
stant derived by considering the partitioning of solutes in saturated
tionary phases as well. West proposes instead that his approach
alkanes was used to compensate for incomplete cancellation of
(not discussed here but developed in his publication) using dif-
dipole induced–dipole interactions, entropy, and other effects. In
ferences in retention indices, which clearly shows the selectivity
these ways, the data treatment involved many steps that are
differences between solvents, correlates better with experimen-
not present in regular solution theory. Thus, while according
tally observed resolution and this provides better predictions and
to theory, dispersion interactions should play a role in solvent
better separations.
selectivity, Snyder took many steps to reduce or eliminate their
In contradiction to West's claims, Snyder et al. [43] cite a presen-
tation given by Starcevic at the 15th International Symposium on
It is also worth examining the work of Meyer and co-workers in
Column Liquid Chromatography (Ref. [23] in the cited work) that
this discussion of dispersion interactions. They quantified the rel-
the selectivities for a different series of compounds did correlate
ative importance of various intermolecular interactions in a series
with predictions from the SST. Snyder et al., however, do not spec-
of papers that examined the cohesive energies (Ec) of polar organic
ify the series, and the authors of the present article did not find any
liquids [46–49]. By examining the densities of polar organic com-
publications by Starcevic to support the claims.
pounds (e.g., 2-ketones) compared to paraffins, the authors wereable to estimate the contributions of orientation (dipole–dipole),
6.2. A note about dispersion
induction (dipole-induced dipole), and dispersion energies to thecohesion of the bulk solvents, defined as "the energy required to
We mentioned above that the SST is based on the assertion that
separate the component molecules to infinity without changing
dispersion interactions in solutions of polar solvents do not con-
the average internal energy of the individual molecules." While
tribute significantly to solvent selectivity. It is important to note
the authors interest seemed to lie more in emphasizing the (some-
that this is very different than saying that dispersion interactions
times overlooked) importance of induction effects, their results are
do not contribute to overall gas/liquid partitioning or chromato-
relevant to our present discussion of the relative importance of
graphic retention. In fact, Snyder used n-alkanes of varying size to
try to remove dispersion interactions in the formation of the SST.
The results for the 2-ketones are shown in Table 2. It is clear that
However, a brief examination of the overall importance of disper-
dispersion accounts for the majority of the interaction energies.
sion interactions is warranted.
For example, for 2-propanone, 71.2% of the cohesive energy arises
Using regular solution theory [44,45], it can be shown that
from dispersion forces. This goes up to over 90% for 2-undecanone.
dispersion interactions do not cancel when considering solvent
Comparable results and trends were observed for n-alkylacetates,
selectivity for gas–liquid partitioning. Specifically, when compar-
n-alkyl nitriles, and 1-chloroalkanes. It should be noted that Kersten
ing the selectivity for two non-polar solutes (e.g., pentane and
and Poole [50] caution that Meyer's methodology is not well estab-
hexane) offered by two different non-polar solvents (e.g., benzene
lished and potentially overestimates the contribution of dispersion
and toluene), selectivity differences between the two solvents exist.
energies to the overall energy of interaction between molecules.
According to regular solution theory, these differences arise from
However, they do not explain why this is so and they acknowledge
(1) differences in the molar volumes of the solutes, (2) difference in
that better alternatives were not available at that time. This caution
the solutes' solubility parameters, (3) differences in the product of
not withstanding, it is reasonable to conclude from Meyer's work
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Table 2
Cohesive energy for a 2-ketones and the percent of dispersion, induction, and orientation forces contributing to the overall energy.
Reprinted with permission from [46]. Copyright 1966 American Chemical Society.
that dispersion interactions play a significant role in retention, even
GC phases fails because the Kovats Retention Index, upon which
for polar solutes in polar systems.
the GC solvent triangles are based, does not account for interfacial
As mentioned earlier in this review, Tan and Carr [42] exten-
adsorption of the test solutes and n-alkane standards and because
sively analyzed the effects of dispersion on retention and how these
of inadequate retention of ethanol, nitromethane, and dioxane (the
effects change as a function of mobile phase modifier and compo-
three probe solutes) on phases of low polarity. After correcting for
sition. They state that the "the contribution of the presumed highly
interfacial adsorption effects (see the publication for more details
unfavorable cavity formation process in water is actually smaller
on how they did this), the authors calculated Xe, Xn, and Xd values
than thought compared to the net favorability of forming disper-
according to the methodology first described by Snyder and plotted
sive interactions with the stationary phase." They then use free
the data as shown in Fig. 14.
energies of methylene group transfer from the gas phase to water
Using a free energy-based parameter,
(+159 cal/mol) and to hexadecane (−634 cal/mol) to indicate the
n, and d, SQ stands for squalane, and PH is the phase of interest,
importance of dispersion interactions to the retention of solutes in
they replotted the data as shown in Fig. 15 [50,51]. The phases
RPLC. They also provide a thorough dissection of the linear solva-
generally shift to the right compared to the plot based on P
tion energy relationships (LSER) that they used to quantify changes
authors attributed this to a decreased contribution from proton-
in the relative importance of dispersion, dipole–dipole, and hydro-
donor forces as measured by the free energy-based parameter and
gen bonding interactions to overall solute retention. They consider
suggested this arises either because none of the phases exam-
their results in light of the amount of water and modifier sorbed
ined have strong proton-donor properties or because dioxane is
into the stationary phase for aqueous methanol, acetonitrile, and
an insensitive probe for measuring proton-donor interactions. In
tetrahydrofuran mobile phases from 20 to 50% (v/v). Overall, they
either case, the authors challenged the basis for the construction of
stress the importance of dispersion interactions between solutes
the SST as applied to GC phases.
and the stationary phase. They also examine the relative cohe-sive energy densities of the mobile and stationary phases whichcontribute to retention via the cavity formation process. Cavity for-
6.4. Further challenges to the SST – test solute selection
mation processes, however, also reflect dispersion interactions inthat interactions between components within the mobile phase or
6.4.1. Kersten and Poole – GC
within the stationary phase must be broken or rearranged in order
Kersten and Poole examined the use of other probe molecules
to create cavities to accommodate solutes. Different organic sol-
[50]. Specifically, they commented on the use of butanol in place of
vents and different compositions will clearly have different effectson the cohesive energy densities of the mobile and stationary phasethat could, depending on their magnitudes, contribute to solventselectivity.
Given the work of Meyer et al. and Tan and Carr, to clas-
sify solvents, it is important to accurately account for dispersioninteractions. Failure to do so may overlook important differencesbetween solvents and their ability to interact with solutes. Thus,if the procedure used by Snyder yields only approximate cancella-tions of dispersion effects, the "excess" dispersion effects must bedistributed (in some unknown fashion) throughout the remainingthree solvent parameters in the SST. This complicates the interpre-tation of these parameters and perhaps also leads to some of theunusual groupings noted in the literature.
6.3. Further challenges to the SST – interfacial adsorption in GC
Kersten and Poole [50] characterized fifteen GC polymeric
phases and found that the relative positions of the phases within
Fig. 14. GC solvent selectivity triangle from Kersten and Poole after correcting reten-
the triangle change depending on the test solutes used to define
tion indices for interfacial adsorption effects.
the apices of the triangle. They further asserted that the SST for
Reprinted from [50], with permission from Elsevier.
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Table 3
Effect of test probes on the Xe, Xd, and Xn values of SE-30.
Ethanol, dioxane, nitromethane
Propanol, dioxane, nitromethane
Butanol, dioxane, nitromethane
Reprinted from [52], with permission from Elsevier.
propanol, and butanol as test probes for SE-30, they found that asthe chain length increased, Xe decreased monotonically and Xd andXn had more complex changes as shown in Table 3. More polarphases such as QF-1 and CW-20M were less affected by the changes.
The authors show that the changes in the position can be
reduced by dividing the corrected retention time of the alcoholhomolog by the corrected retention time of its corresponding n-alkane. However, the authors also noted changes in positions when
Fig. 15. GC solvent selectivity triangle calculated using the free energy-based
nitromethane was replaced by acetonitrile or nitropropane as the
parameter, ı(�G0)
, to define the sides of the triangle.
polarity indicator. Normalizing for changes such as switching from
Reprinted from [50], with permission from Elsevier.
nitropropane to acetonitrile would be more difficult as there isnot an underlying homolog series in common as there is for then-alcohols.
ethanol, nitropropane in place of nitromethane, and 2-pentanone
The success of the normalization procedure suggests that the
or pyridine in place of dioxane. They provided evidence that chang-
number of methylene units in the test compounds is important,
ing the three probe solutes would change the position of the GC
which in turn suggests that dispersion or induction effects are not
phases within the triangle in an unpredictable manner. Unfortu-
being completely removed by subtracting the retention index on
nately, they do not actually replot the selectivity triangle based on
squalane. It should be further noted that in this study, no attempt
these new probes to visually demonstrate the changes in position.
was made to account for solute interfacial adsorption or inadequate
Changes in position, particularly if groupings change as a result,
retention of the test molecules. Kersten and Poole demonstrated
would call into question the utility of the SST approach as a means
that this can alter retention indices and consequently X
of classifying solvents. They also perhaps reinforce the concerns of
i values [50].
Some of the observed changes with increasing probe chain length
West examined earlier.
may therefore be due to changes in the relative contributions ofadsorption and absorption (i.e., partitioning) to the retention of the
6.4.2. Shah, Na, and Rogers – GC
probes and the n-alkanes.
Shah et al. had earlier noted the sensitivity of the position of GC
phases within the triangle to the choice of test solutes [52]. They
6.4.3. Betts – GC
used Klee's definition of Xi values (defined earlier in this review)
Betts also published a GC triangle using yet another set of probe
and characterized the same six phases. Fig. 16 shows a comparison
solutes [53]. Based on his work, he ultimately recommended that
of the results from the two reports. In general, the agreement is
three GC phases are essentially all that are required for most sep-
arations (SE-30, polysiloxane; QF-1, trifluoropropyl; and XE-60,
When butanol rather than ethanol was used as the test probe,
cyanoethyl) – three that had been identified in 1969 as being among
the position of the phases changed dramatically. For example,
the most used phases around that time, 15 years before Betts pub-
the value of Xe for SE-30 changed from 0.534 to 0.246, which
lished his findings [54]. Betts' somewhat vehement response to
clearly would change its grouping. By examining the series ethanol,
Klee et al.'s prediction that a computerized optimization for mak-ing new mixed GC stationary phases would eventually be in place[14] was "There are already far too many; let us not mix them!"Betts also cites McReynolds, who, based on his own work, wrote"It is hoped that this data will help reduce the number of liquidphases being used" [12]. He said this because of his finding thatmany phases show similar characteristics. It would be interestingto hear Betts' thoughts on today's era of two-dimensional GC andLC separations (which in some ways can be [incorrectly] thoughtof as mixed phases of a sort) and the hundreds of commerciallyavailable LC and GC phases.
6.4.4. Cooper and Lin – RPLC
Based on Snyder's selectivity triangle, Cooper and Lin [55]
selected toluene, phenol, aniline, and nitrobenzene to test the rel-ative importance of proton donor, proton acceptor, and dipolecharacteristics of RPLC mobile and stationary phases. Toluene wasused as a reference compound and the slopes of plots of ln k vs.
volume fraction of organic modifier obtained with toluene were
Fig. 16. Selectivity triangle comparing locations of six GC stationary phases reported
subtracted from comparable slopes for the other compounds. The
by Klee et al. [14] with those from Shah et al. [52] to ascertain reproducibility ofpositions within the triangle using ethanol, dioxane, and nitromethane as probe
intention was to isolate just the retention of the functional groups.
solutes to determine X
In some ways, this is similar to Snyder's approach of correct-
e , Xd , and Xn , respectively.
Reprinted from [52], with permission from Elsevier.
ing partition coefficients of solutes by subtracting the partition
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
coefficient of an alkane of the same size. Unfortunately, the authors
tive effects make the behavior of these two solutes different enough
do not convert their findings into values that can be plotted in a
so as to provide distinct chemical information.
triangle and thus the results of this study using different probesolutes cannot be readily compared to other studies using Snyder's
7. Re-evaluation of the SST using solvatochromism and
linear solvation energy relationships (LSERs)
7.1. Reevaluating the SST using solvent solvatochromic
6.4.5. Smith – RPLC
The last paper we will note in this section regarding Snyder's
selection of probe solutes is that of Smith [56]. In this work, prin-
In 1989, Snyder participated in a reevaluation of the solvent
cipal components analysis of RPLC data on eight columns with
triangle [57]. This work produced three major results:
three different mobile phase compositions was used to determinethe number and identity of test compounds needed to account forthe variance in retention indices using alkyl aryl ketones as stan-
(1) More thermodynamically rigorous corrections for dispersion
dards. Six potential test solutes were studied (aromatic analogs
and entropy (cavity formation) effects produced only slight
of Rohrschneider's and McReynold's standards). Smith ultimately
modifications to the relative position and groupings of solvents
concludes that toluene, nitrobenzene, 2-phenylethanol, and p-
compared to the original SST, and the modifications that did
cresol give optimal discrimination between mobile and stationary
result could be rationalized chemically,
phases. This is an interesting result in that four compounds were
(2) The selectivity parameters (Xe, Xd, and Xn) were shown to be
found to be necessary, in contrast to Snyder's suggestion that three
composite values comprised of dipolar, hydrogen bond acidity,
suffice. It is also interesting that toluene is included in the four. This
and hydrogen bond basicity effects, and
suggests that dispersion and/or dipole–induced dipole interactions
(3) The three original probe solutes used to develop the SST were
are important in discriminating/characterizing different mobile
acknowledged to be "inefficient" choices in terms of their ability
and stationary phases, which is consistent with Meyer et al.'s work
to discriminate between solvents.
discussed above. If this is correct, then mobile or stationary phaseswith greater ability to participate in these interactions could show
We will leave the interested reader to explore points 1 and 3 in
greater selectivity for non-polar and polarizable compounds. This
the publication and focus on the second point.
seems to be an argument against Snyder's assertion that disper-
To analyze the meaning of the selectivity parameters, the
sion effects are negligible in solutions of polar solvents [10]. Snyder
authors plotted values of Xe, Xd, and Xn for various solvents vs. the
does acknowledge that "there is no doubt that the inclusion of
solvent parameters ˇ, ˛, and �*, respectively. The parameters �*, ˛,
additional test solutes improves the ability to predict solute reten-
and ˇ, are measures of solvent dipolarity/polarizability, hydrogen
tion behavior and to carry out fine-tuning of the solvent selectivity
bond donating ability, and hydrogen bond accepting ability, respec-
based on second-order effects" but doing so "appears more to con-
tively [58–62]. They are derived largely from spectroscopic shifts
fuse than to clarify our understanding of solvent selectivity for
of aromatic compounds that are sensitive to their chemical envi-
a given application." Snyder does continue on in his article to
ronment and hence are sometimes referred to as solvatochromic
consider X values for toluene and shows them to have little vari-
parameters. Given that they are based on spectroscopic data, they
ation between different solvents. One could postulate, then, that
are derived from data entirely independent from that used to define
in Smith's study, toluene would be the least important of the four
P� and Xi values. Furthermore, through judicious selection of mul-
probes in terms of its contribution to the primary principal compo-
tiple solvatochromic probes, the �*, ˛, and ˇ scales were very
nents that recreate the data set and that it is there to ‘fine-tune' the
carefully constructed to measure only the solvent interaction abil-
model for minor effects of dispersion forces. This is not the case,
ity of interest and to exclude contributions from other possible
however, as shown by the principal components analysis results
interactions (e.g., ˛ is a measure of just a solvent's HB donating
ability with very little or no contribution from polarity or HB accept-
It would be interesting to see the effects of dropping toluene
ing ability). It is also worth noting that dispersion interactions
from the data that was fed into the principal components analysis.
play almost no role in solvatochromism. Dipole-induced dipole
Unfortunately, Smith did not perform this analysis. It is also inter-
effects arising from solvent polarizability do contribute, though,
esting to note the apparent redundancy of 2-phenylethanol and
and dispersion interactions tend to be collinear with polarizabil-
p-cresol as test probes, which suggests that resonance and induc-
ity. Dipole–induced dipole interactions tend to be much smaller
Table 4
Factors calculated with principal components analysis of the retention indexes of six reference compounds.
Contribution from reference standard
Reprinted with permission from [56]. Copyright 1984 American Chemical Society.
a Percent of overall variance attributed to this component. Components 4–6 have been omitted because they contribute so little (<4%) to overall variance.
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
in magnitude than dispersion interactions, as indicated by Meyer'swork discussed above.
Plots of Xi values for a variety of solvents vs. their corresponding
�*, ˛, or ˇ values are shown in Fig. 17 [57]. Assuming �*, ˛, ˇ tobe ‘pure' scales in terms of measuring only a single intermolecularinteraction ability (which is known not to be case for �*), if Xi valuesfor a set of solvents correlate strongly with the corresponding sol-vatochromic parameters, then the Xi values would also be said to bequite pure. However, as the authors point out, there is "disappoint-ingly little correlation of the selectivity factors with the individualsolvatochromic parameters." They note, however, that in reality,it is not the Xi values that should be correlated with the solva-tochromic parameters, but rather P�Xi values. Still they found littlecorrelation between parameters suggesting that the two methodsof characterizing solvents were not measuring the same attributes.
To understand better what the Snyder parameters represented, theauthors next used multi-parameter linear regressions to correlateP�Xi with the solvatochromic parameters �*, ˛, ˇ, and ı, where ı isa term added to account for polarizability effects not incorporatedin �* [63]. The correlations thus took the form
P�Xi = SPo + s�∗ + dı + a˛ + bˇ + h˛ˇ
where SPo is an intercept term. The parameter ı is set equal to 0.00,0.500, or 1.00 for aliphatic, halogenated, and aromatic solvents,respectively. The results of the regression are shown in Table 5.
As the authors point out, these results indicate that all three
original test solutes have appreciable dipolar interactions withsolvents as indicated by their large positive s-coefficients. Further-more, dioxane and ethanol are both sensitive to solvent hydrogenbond acidity (positive a-coefficients), negating the assumption thatXd is the primary measure of solvent HB donating ability in theSST. It was also concluded that the assumption that ethanol is themain probe of solvent basicity is correct as evidenced by its largeˇ-coefficient compared to that for dioxane and nitromethane. Inthis way, the authors showed that
(1) Xi is a composite of solvent dipolarity/polarizability, HB acidity,
(2) Xd reflects a blend of solvent dipolarity and HB acidity, and(3) Xn mainly reflects dipolarity with smaller contributions from
HB acidity and basicity.
The authors go on to propose that triethylamine (�* = 0.14,
˛ = 0.00, ˇ = 0.71) and trifluoroethanol (�* = 0.73, ˛ = 1.51, ˇ = 0.00)be used to probe solvent HB acidity and basicity, respectively. This
Fig. 17. Plots of Snyder parameters vs. related Kamlet–Taft solvent parameters to
is based on their relatively high ˇ/�* and ˛/�* ratios. While the
compare the similarity (or lack thereof) of solvent properties measured by each. (a)
˛-value for trifluoroethanol is high, a �* value of 0.73 is also quite
Xe vs. ˇ, (b) Xd vs. ˛, and (c) Xn vs. �*.
Reprinted from [57], with permission from Elsevier.
high (given that the scales generally range from 0.00 to 1.50), suchthat it is questionable as to how much using this probe would helpin determining a pure basicity contribution to the SST free fromdipolar interactions.
Table 5
LSER coefficients showing the contributions of polarity, dispersion, and hydrogen bonding effects on the P�Xi values of several probe solutes.
Reprinted from [57], with permission from Elsevier.
Standard deviations of the coefficients are in parentheses.
a These coefficients were found to be not significantly different from zero and were omitted in the final fit.
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Fig. 18. Solvatochromic parameter-based plot of properties of aliphatic solvent classes and some specific solvents (left). Further simplification of the plot obtained by
averaging Xi values for solvents of a given type (amines, alcohols, etc.) (right).
Reprinted from [43], with permission from Elsevier.
7.2. Solvatochromic SST for solvents
explain the exact nature of the dependence of the original SST onprobe solute selection. The use of solvatochromic parameters to
As an extension of the ideas presented in the article just dis-
reconstruct the SST was also new.
cussed, Snyder et al. reconstructed the SST using the Kamlet–Taftsolvatochromic parameters to define the apices [43]. To do so, each
7.3. Solvatochromic SST and practical RPLC considerations
solvent parameter (�*, ˛, ˇ) was normalized to the sum (
It is worth extending our discussion of Snyder et al.'s exami-
the three parameters for each individual solvent. The normalized
nation of the SST as it relates to solvent selection in RPLC [43]. As
parameters were used as the apices of the triangle.
presented earlier, the SST was used as the basis for many optimiza-
In order to facilitate the comparison between the solva-
tion schemes. However, while the SST was proposed as a guide to
tochromically based triangle and Snyder's original SST, the authors
aid in solvent selection, it does not provide guidelines regarding
averaged Xi values for solvents within a class (e.g., amines, alcohols,
the effect of increasing the percent water in each of the solvents on
etc.). They also only compared aliphatic solvents, largely because
chromatographic selectivity. Along those lines, the authors noted
all but one of the aromatic solvents studied had ˛ = 0.00 and thus
that the "SST approach assumes that solvent strength can be varied
they clustered along the right vertex of the triangle. The original and
(by varying the percent water in RP-HPLC) without changing selec-
‘new' plot, thus simplified, are shown in Fig. 18 [43]. The authors
tivity" (italics ours). But water is certainly not a passive diluent, as
the authors attest by pointing to changes in the �*, ˛, ˇ values of
"the relative positioning of different solvents. .is similar in that
solvents modified with varying amounts of water [30,31,64,65]. We
solvents which are more basic, acidic, or dipolar in [the original]
agree with their statements that the "SST approach to adjusting sol-
are also more basic, acidic, or dipolar [in the new plot]. A further
vent strength and selectivity in RP-HPLC is overly simplified" and
examination. .however, shows that solvents of similar acidity
that "it is all but impossible to vary the mobile phase strength via
or basicity are better grouped in the solvatochromic approach.
a change in the water content without also changing some other
Thus, amines and ethers show up as distinctly basic, as com-
significant solvent-selectivity property" [43]. Specifically, they go
pared to the alcohols in [the original plot]. The alcohols, glycols,
on to note that previous studies show that, with respect to selec-
formamide, carboxylic acids, water, and chloroform show up
tivity, water simultaneously affects the mobile phase cohesivity
as acidic solvents in [the new plot]. The acidity of these latter
(i.e., the ease of cavity formation to accommodate solutes), polarity,
solvents seems inadequately expressed in [the original]."
and HB acidity, with only minor changes in basicity. They suggest,therefore, that for RPLC purposes, the SST should be reconstructed
They go on to suggest that these problems (and a few oth-
using surface tension, or some other cohesivity-related property,
ers) in the original SST can likely be attributed to the fact that
polarity, and hydrogen bond acidity to define the apices, thus elim-
nitromethane, ethanol, and dioxane do not provide pure mea-
inating hydrogen bond basicity. The fact that the mobile phase
sures of polarity, basicity, and acidity, whereas the solvatochromic
also modifies the stationary phase properties is also noted as a
parameters were designed to do just that. Furthermore, the solva-
complication in predicting selectivity changes between different
tochromic parameters are averages obtained with several probes
solvents and solvent compositions. These effects were comprehen-
for the determination of each �*, ˛, and ˇ value. This reduces some
sively explored by Tan and Carr [42]. Using linear solvation energy
of the probe-specific effects that are inherently embedded in the
relationships, they demonstrate changes in the relative contribu-
construction of the SST.
tions to solute retention of cavity formation, dispersion, dipolarity,
As a final, and rather eloquent, explication of why it is that
and hydrogen bonding interactions as a function of mobile phase
the original three probes represent blends of interactions, theauthors provide their solvatochromic parameters, reproduced herein Table 6 [57]. These values make it clear that all three solutes are
polar and that dioxane and ethanol are both good hydrogen bond
�*, ˛, and ˇ values of Snyder's original test probes showing that they participate in
acceptors (high ˇ values). Thus, any parameters derived from this
a blend of intermolecular interactions and do so to varying extents.
triad of solutes will necessarily represent blends of interactions.
Given all of the challenges that had come before this work, the
solute-dependent nature of the original SST was not a novel reve-
lation (as the authors acknowledge via their citations). What was
new, however, was the use of the solvatochromic parameters to
Reprinted from [57], with permission from Elsevier.
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
modifier (methanol, tetrahydrofuran, and acetonitrile) and com-position. They review multiple studies of sorption of water andorganic modifier into the stationary phase [66–75] and interprettheir findings in light of the modifications of the stationary phaseby sorbed water and organic solvent. Snyder et al. [43] acknowl-edge that if these changes were incorporated into an LC-specifictriangle, the solvent classifications/groupings would change, butthey defend the overall SST approach as a useful one in a qualitativesense.
7.4. Solvatochromic SST for GC
Li et al. extended the considerations of solvatochromically
derived LSERs to include characterizing GC phases [76]. Theirapproach is based on the LSER equation
where SP is the logarithm of the specific retention volume (Vog),partition coefficient (K), or retention factor (k) for a compound(denoted by the number ‘2') on a given phase. Log L16 is the solutegas-to-n-hexadecane partition coefficient. The parameters �∗,c, ˛c ,
and ˇc are measures of a solute's dipolarity/polarizability, hydro-
Fig. 19. Classification of GC stationary phases using a triangle based on LSERs. See
gen bond acidity, and hydrogen bond basicity, respectively [77,78].
reference for groupings.
The ı2 parameter is meant to account for polarizability effects not
Reprinted with permission from [76]. Copyright 1992 American Chemical Society.
included in �∗,c. SP
o is a solute-independent constant specific to
the stationary phase being studied, and l, s, a, b, and d are column-
than just three. The second is that each of the probe solute's sol-
related parameters determined by regression of SP measured for
vatochromic parameters are more carefully defined to isolate the
a large number of solutes (dozens if not hundreds) against their
specific intermolecular interactions being represented, as opposed
corresponding �∗,c, ˛c , ˇc , and ı
2 values. The important aspect
to Snyder's three probe system which was shown earlier to have
is that the coefficients quantify the ability of the stationary phase
blends of interactions represented by each probe. This study is
to interact with solutes through various intermolecular forces. For
also interesting because of its redefinition of the apices to include
example, the ˛c parameter reflects the solutes' abilities to donate
dispersion effects (embedded in the l log16 term) in favor of HB
hydrogen bonds. Therefore, a large, positive a-coefficient indicates
donating effects, which seems an imminently reasonable substi-
that the stationary phase strongly retains HB donors and is there-
tution based on the actual intermolecular interactions that govern
fore itself a strong HB acceptor (i.e., the phase is basic). Thus, the
selectivity in gas chromatography.
coefficient reflects the complementary property of the solute.
The authors selected 53 representative GC phases from data
7.5. An important note about LSER ratios
sets collected by McReynolds [13], Poole and co-workers [79], andCarr and co-workers [77,80] and performed LSER analyses accord-
While Li, Zhang, and Carr based their solvatochromic GC trian-
ing to the equation above. Principal components analysis showed
gle on absolute values of the LSER coefficients, later work from the
that three components account for over 98% of the variance in the
same research group indicates that for determining selectivity, the
Poole and McReynolds data sets (results for Carr's data set were
ratios of the LSER coefficients are the distinguishing parameters
not provided). Based on this and the LSER results, it was con-
[81]. This can be shown by considering the correlation of log k val-
cluded that three parameters could be used to characterize each
ues with two independent solute properties (Xi and Yi) as shown in
GC phase. Because the l, s, and a coefficients (dispersion/cavity for-
the equations below (LSERs generally use four or five parameters,
mation, dipolarity/polarizability, and HB acidity) have the largest
but considering two here suffices to illustrate the point):
LSER coefficients, those coefficients were selected to define theapices of a solvatochromically based GC–SST. There are very few
log k1,i = a1 + b1Xi + c1Yi
GC phases that are even slightly good hydrogen bond donors, so the
b-coefficients are always quite small or statistically insignificant.
2,i = a2 + b2Xi + c2Yi
As in the earlier solvatochromic study, the coefficients had to be
The subscripts 1 and 2 refer to two different chromatographic
normalized in order to be plotted in a triangle. Three parameters
systems (e.g., two different stationary phases, mobile phases, or sta-
were thus defined:
tionary/mobile phase combinations). Combining the two equationsyields
Note that if the ratio c1/b1 = c2/b2 then the retention on one phase
correlates perfectly with retention on the other, regardless of what
solutes are used or their properties. Zhao and Carr state that in this
The resulting triangle plot is shown in Fig. 19 [76]. The authors
case, there is no difference in the "effective selectivity" of the two
comment that very few phases are located in the HB acceptor
systems. By "effective selectivity" they mean differences that lead
corner owing to the fact that few columns are very basic while
to elution order changes and differential changes in band spacing, as
being of low polarity. They also reiterate two advantages of an
opposed to merely spreading out peaks a little more in one system
LSER-based triangle scheme for phase classification. The first is
compared to another. If c1/b1 /
= c2/b2, then retention on the two
that LSERs are determined using dozens of probe solutes rather
phases might not be correlated and instead depends on the solute
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
properties present in the analyte mixture. In this case, if both Xiand Yi for the solutes make substantial contributions to retention,then "effective" or useful changes in selectivity could result fromchanging from one system to the other. So the key to obtaininguseful selectivity is to have ratios, not necessarily absolute mag-nitudes, of LSER coefficients that are different. For this reason, theGC selectivity triangle discussed previously would have been bet-ter presented if ratios of LSER coefficients had been used to definethe apices rather than absolute magnitudes.
8. Recent uses of selectivity triangles – MEKC, RPLC, and
NPLC
8.1. MEKC selectivity triangles based on LSERs
Based on all of the work over the years that has been summa-
rized above, Fu and Khaledi have recently characterized pseudophases used in electrokinetic chromatography (EKC) [82,83]. Thephases included elution buffers modified with micelles, polymers,
Fig. 20. Micellar selectivity triangle based on LSERs. See reference for groupings.
vesicles, liposomes, mixed micelles, polymer/surfactant mixtures,
Reprinted from [82], with permission from Elsevier.
and organically modified pseudo phases. This is the first report thatthe present authors are aware of in which an SST approach wasapplied to micellar and related systems.
As a critical test of the methodology and the reproducibility of
Their triangle is ultimately based on LSERs of the form
using LSERs in this manner, Fu and Khaledi collected fourteen dif-ferent literature reports of LSERs for sodium dodecyl sulfate (SDS)
and plotted them in a triangle shown in Fig. 21. It is reassuring thatthey generally cluster together. Reasons that a few of the results
where V, B, A, S, and E are measures of a solute's volume, HB basic-
are outliers can be offered based on differences in experimental
ity, HB acidity, dipolarity/polarizability, and excess polarizability,
respectively [84,85]. The values of c, v, b, a, s, and e are determined
Returning our attention to Fig. 20, the authors identified four
by linear regression analysis and were either taken from literature
different groupings of systems labeled A, B, C, and D. As with other
reports or measured by the authors.
selectivity triangles, the suggestion is that if a system in one group-
The definition of the apices of their triangle is somewhat more
ing does not achieve the desired separation, then switching to
complex than others discussed in this review. First, the authors
another system within the same group is unlikely to produce dra-
calculated the ratio of LSER coefficients as suggested by the Carr
matic changes in elution order or selectivity. Rather, it would be
publication discussed above [81]. They choose to normalize to the
better to change to a system in a different group that might have
v-coefficient, which is commonly done because the v-coefficient is
different blends of intermolecular interactions and therefore might
often one of the largest in LSERs, particularly those involving solute
offer different selectivity.
transfer into or out of aqueous phases. The symbol Ii is given to eachpossible ratio (a/v, b/v, s/v, e/v), where the subscript ‘i' representsany one of the individual ratios. Next, the I
8.1.1. Analysis of the MEKC selectivity triangle
i values are converted to
As with the original Snyder triangle, some challenges can be
made to this classification scheme. First, the vertices are labeled
with "polarity", "basicity", and "acidity" as if they are absolute mea-
where the ranges of Ihigh and Ilow were selected to incorporate (butnot equal) the high and low values for each ratio for the 74 sys-tems studied. For example, Ihigh = −1.50 and Ilow = 0.00 for b/v, eventhough for the 74 systems, the actual lowest value was −1.38 andthe highest was −0.23. While the actual Ihigh and Ilow values wereadjusted for each ratio being considered, the difference betweenthem (i.e., the range) was kept constant at 1.50. Defining Ui in thisway provided quantities that were all positive (unlike LSER coeffi-cients which can be positive or negative) and which ranged from0.00 to 1.00.
Next, in a manner similar to that in Brown's original work, the
Ui values were normalized according to
such that Xa + Xb + Xc = 1.00. These three Xi values then served asthe apices of the micellar selectivity triangle (MST). With four LSERcoefficient ratios, there are obviously several different triads thatcan be used to define a triangle and the authors explored theseoptions. They ultimately recommended one based on Xb + Xs + Xa.
Fig. 21. Micellar selectivity triangle showing data points based on fourteen litera-
Using this scheme, the 74 systems were plotted as shown in Fig. 20
ture reports for sodium dodecyl sulfate LSERs.
Reprinted from [82], with permission from Elsevier.
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Fig. 23. Plot of b/v vs. b to show the influence of v-coefficients on the interpretation
of pseudophase properties as defined by I-values in the development of the MST.
Fig. 22. Plot presented to show a poor correlation between Xb and v-coefficients,
ascending order. This suggests that the smaller v-coefficients (per-
suggesting that the X scales in the MST are not influenced by differences in
haps arising from easier cavity formation or weaker solute/micelle
v-coefficients for the various pseudophases.
Reprinted from [82], with permission from Elsevier.
dispersion interactions in these micelles compared to others) areleading to the high magnitudes of b/v ratios. Thus, it is perhaps notan acidity effect but rather a different effect that places them inthe group in which they reside within the triangle. This suggestion
sures, whereas in reality they are quite complex parameters. Their
is supported by arbitrarily replacing the actual v-coefficients for
complexity can be recognized by first considering that they are
these systems with the average v-coefficient calculated using all of
ratios of LSER coefficients. Then, they are normalized to an arbi-
the systems. If the plot is remade (Fig. 24), it is clear that the eight
trary high and low value, and then get renormalized to the sum
‘outliers' fit into the general correlation. So the apparent enhanced
of three such normalized parameters. This means that they are
magnitude of the AGENT and Elvacite 2669 systems (in terms of
not truly direct measures of any single selectivity characteristic of
larger negative b/v ratios) is more a result of low v-coefficients.
the system. For example, when the authors state that "Group B is
Similarly, the apparently smaller-than-expected magnitude of the
mainly comprised of fluorinated micelles and could be considered
b/v ratio of octanol/water partitioning is a result of a larger-than-
the strongest hydrogen bond donor and weakest hydrogen bond
average v-coefficient, as demonstrated by replacing its v-coefficient
acceptor among all the micelle systems" or "In general, the compo-
with the average and replotting it as shown in Fig. 24. These con-
nents in group C are slightly stronger hydrogen bond acceptors and
cerns do not negate the use of the MST, but they complicate the
weaker hydrogen bond donors than those in group A", these state-
chemical understanding of why the systems fall where they do
ments would only be true if the v-coefficients for all of the systems
within the triangle.
were the same. However, the v-coefficients in their collection vary
In terms of absolute strengths of interactions, it is useful to look
from 1.49 to 3.78 (excluding the value of 3.94 for octanol/water par-
at the interpretations of LSERs. In general, the solute partitioning
titioning which was included in their study as a bulk phase model of
is defined as the transfer of the solute from the aqueous phase into
micelle/water partitioning). This is a relatively large range for LSER
the pseudo phase. Thus, negative coefficients indicate the solutes
coefficients and quite comparable to the overall range for the b-
partition less as their solute parameters increase. For example, a
coefficients in this study (−0.47 to −3.86). Even if the v-coefficients
negative b-coefficient indicates that the aqueous phase is a stronger
were the same, it is difficult to make these statements because Xi is
hydrogen bond donor than is the pseudo phase (which typically
a relative measure of the property under consideration compared
makes sense given the ability of water to donate hydrogen bonds).
to the sum of three different properties. Thus, all that really can
The magnitude of the coefficient indicates the degree to which this
be said, for instance, is that the contribution of the acidity/cavity
is true. The larger the magnitude, the weaker the hydrogen bond
formation ratio compared to the overall sum of basicity/cavity for-
donating ability of the pseudo phase. A coefficient of zero indicates
mation, acidity/cavity formation, and polarity/cavity formation is
that the pseudo phase is just as strong a donor as the aqueous phase,
highest for systems in group B. Even this, however, is an oversim-
and a positive coefficient indicates that the pseudo phase is a better
plification because the solute parameter V (solute size) models bothcavity formation (endoergic) and dispersion (exoergic) effects. Sothe interpretation of the v-coefficient is itself not simple. Thus, atbest, the descriptors along the sides of the triangle are oversimpli-fications and serve, perhaps, as first approximations or convenientlabels of what are quite complex measures of the systems'characteristics.
To demonstrate that the X scales are not influenced by the
magnitudes of the v-coefficients in the manner suggested in theparagraph above, Fu and Khaledi show a plot of Xb vs. v, which hasgenerally scattered data for the 74 systems (see Fig. 22) [82].
We have developed the related plot of b/v vs. b shown in Fig. 23.
If all of the v-coefficients were identical, such a plot would resultin a straight line. However, it is clear that several systems fall wellbelow the general correlation. The outliers correspond to the sevenAGENT polymeric micelles and Elvacite 2669. These eight systems
Fig. 24. Same plot as in the previous graph except that the v-coefficients for the
have the six lowest v-coefficients of all 74 systems and the other two
AGENT surfactants, Elvacite 2669, and octanol–water have been replaced with the
are numbers nine and twelve when all 74 v-coefficients are listed in
average v-coefficient for the set of 74 pseduophases.
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Fig. 25. Plots of Xi vs. i to compare the triangle parameter (Xi) to the LSER (i) parameter and the complications that can arise when interpreting Xi values. (a) Xb vs. b, (b) Xa
vs. a, (c) Xs vs. s.
HB donor than the aqueous phase. When one lists the b-coefficients
they actually interact more strongly with polar solutes than does
for all of the systems from smallest negative (i.e., strongest donor)
water (or more precisely, the aqueous phases used in these studies).
to largest negative (i.e., weakest donor) the perfluorinated surfac-
Thus, it appears that the normalization process is not accurately
tants are numbers 1–5 and 11 in a list of all of the systems. Thus,
simultaneously reflecting all of the properties of these phases.
on the absolute scale, they are the strongest HB donors just as they
As a final illustration of the problem of interpreting the axes,
are in the triangle, so the normalization process does not appear to
when the authors plot Xb + Xs + Xe (instead of Xb + Xs + Xa) the per-
distort their position with regards to their HB donor strength. How-
fluoro surfactants switch from having the lowest ‘dipolarity' values
ever, the AGENT pseudo phases are seen as some of the weakest
to the highest (figure not shown).
HB acids on the triangle, which is inconsistent with their absolute
Of course, it must be noted that some of the above conclusions
values when compared to all of the other systems.
about the MST are drawn based on a consideration of the absolute
It is possible to argue that the normalization of b/v values to
values, and not ratios of LSER coefficients which, as pointed about
high and low values and then to the sum of Ui values as dictated
above, are the more critical parameters to consider when interested
by the methodology removes the effect of the small v-coefficients
in selectivity differences between systems. Nevertheless, while they
such that the Xi values that are ultimately plotted in the triangle
do not negate the utility of the triangle, they do show that the state-
accurately reflect the relative intermolecular interaction strengths
ments made about the various systems and the labeling of the sides
of the mobile phases. Fig. 25a–c shows plots of Xi vs. I for i = b, a,
of the triangle are at best oversimplifications.
and s. These plots make it clear that the way in which the apicesof the triangle are defined, combined with the magnitude of the
8.2. RPLC column selectivity triangle based on the hydrophobic
v-coefficients, can produce over- or underestimated strengths of
subtraction model
specific classes of phases. For example, the AGENT surfactants haveunderestimated Xb and Xs values given their absolute magnitudes
Quite recently, Zhang and Carr [86] published multiple trian-
for the corresponding b- and s-coefficients. These underestimated
gles based on the Snyder–Dolan hydrophobic subtraction model of
Xb and Xs values are offset by overestimated Xa values. These values
column selectivity [87] which takes the form
do not result because of enhanced acidity and polarity of these sur-
factants, but rather because of their small v-coefficients relative to
= ��H − ��S∗ + ˇ�A + ˛�A + ��C
most systems. The fact that their basicity (X
a) is overestimated is a
mathematical artifact that arises from the requirement for the sum
where the column parameters H, S*, A, B, and C are obtained
of all X values to equal 1.00. If some parameters are underestimated,
via multiparameter linear least squares regression of log(ki/kEB)
then others will necessarily be overestimated.
against the known solute descriptors ��, ��, ˛�, ˇ�, and �� for a
To further complicate the analysis of the triangles, we note that
set of solutes, i, analyzed with a given mobile phase and sta-
group B is seen to have the lowest dipolarity according to the labels
tionary phase for different columns. H, S*, A, B, and C provide
on the triangle. This contradicts what is observed based on absolute
measures of solute–column interactions. Specifically, they rep-
values of the s-coefficients. In fact, the perfluoro surfactants are the
resent hydrophobicity, steric resistance, HB acidity, HB basicity,
only pseudo phases that have positive s-coefficients, indicating that
and cation-exchange activity, respectively, of the mobile/stationary
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Fig. 26. RPLC selectivity triangles based on the Snyder–Dolan hydrophobic subtraction model for 366 stationary phases and a single mobile phase with a particular weighting
factor (see reference for details). (a) S*–B–C triangle, (b) S*–A–C triangle, (c) A–B–C triangle, (d) S*–A–B triangle.
Reprinted from [86], with permission from Elsevier.
phase combination being studied. Data for sixteen solutes analyzed
shown in Fig. 26. These triangles used a weighting factor defined
on 366 commercial RPLC phases were collected and analyzed using
in such as way as to yield the same quantitative effect on phase
the hydrophobic subtraction model. The resulting H, S*, A, B, and C
selectivity (defined as the standard error in a log k vs. log k plot for
values were used to construct selectivity triangles according to the
retention of sixteen solutes on the different phases) for an equiva-
lent numerical change in two different normalized phase properties
First, ratios of the coefficients were calculated (in accord with
(see the original reference for more details). A brief examination
the earlier discussion which showed that it is the ratios, not the
of the triangles developed using this weighting shows that the C
absolute values of coefficients, that must be compared in order to
parameter (ionized silanol effects) dominates the three triangles in
compare the selectivities of two different chromatographic sys-
which it appears. The effect is to cluster all but the most dissimilar
tems). The authors selected H as the parameter to which other
phases. Such clustering makes it impractical to use the triangles to
coefficients were normalized.
select phases of differing properties, or to distinguish one group
The authors then defined a parameter Xi as
of columns from another as is usually done with triangles. Theauthors thus sought a different weighting scheme, using instead
Xi = (I − Imin)�i
where I = S*/H, A/H, B/H, or C/H and �i is a weighting factor. Todevelop a triangle, three �
j values were defined as
Such that Xi becomes
where j = S*, B, or C.
Clearly, with four different I-ratios, four different sets of three
I-values are possible. Thus, four different triangles were plotted as
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Fig. 27. RPLC selectivity triangles based on the Snyder–Dolan hydrophobic subtraction model for 366 stationary phases and a single mobile phase with a different weighting
factor compared to the previous plot (see reference for details). (a) S*–B–C triangle, (b) S*–A–C triangle, (c) A–B–C triangle, (d) S*–A–B triangle.
Reprinted from [86], with permission from Elsevier.
which is akin to one of the steps in the development of the micellar
they may not be truly needed as they do not add to our ability
selectivity triangle (MST) of Fu and Khaledi discussed elsewhere in
to achieve separations. Their second, and perhaps more impor-
this review.
tant conclusion, is that commercially available stationary phases
The use of this weighting factor resulted in the four selectivity
are not exploring all possible blends of intermolecular interactions
triangles shown in Fig. 27. With these triangles, the authors find that
and thus not providing a full range of selectivities. It is interesting to
type-B silicas derivatized with alkyl chains are generally grouped
note that this is the same conclusion that Brown reached 50 years
together in the center. Phases derivatized with cyano, phenyl, flu-
ago, and which Betts reiterated in 1986, regarding GC stationary
oro, or polar embedded groups and those based on type-A silicas
phases as discussed above.
generally show larger differences in coefficients and fall outside ofthe central cluster. All phases of a given chemical compositional
8.2.1. Analysis of the RPLC selectivity triangle
class certainly do not fall in the same region of the triangle.
Given the similarity of this approach based on the hydropho-
The authors also comment on some surprising findings. For
bic subtraction model with that of Fu and Khaledi's based on LSERs,
example, the three chemically different columns (ACE AQ – a polar
many of the same potential advantages and disadvantages exist. For
embedded phase; Betasil Phenyl-hexyl – a phenyl phase, and Bond-
example, both approaches rely on entire sets of solutes, as opposed
clone C18 – a type A alkyl silica) are near each other in the triangle.
to just three probe solutes, to define the apices of the triangle. This
This suggests that their selectivities are comparable, as was verified
increases the probability that the results are more broadly repre-
by high correlation coefficients and small standard error values for
sentative and would apply to a broad range of solutes.
regressions of log k on one column vs. log k on another for the set
A disadvantage is that to create the plots, one parameter of the
of sixteen variegated test solutes.
model is ignored. Thus, two phases that are identical in three of the
Zhang and Carr make two important points about the phases
four parameters may appear to have similar selectivities in one of
they studied, stating that "a huge fraction of the available space [in
the triangles. Therefore, use of any one triangle may be blind to a
the triangles] is under populated and certain regions are extremely
major difference in selectivity of two phases which is very evident
over populated" [86]. So their first conclusion is that on one hand,
in a different triangle. This is particularly important if the solute
many of the phases are quite comparable to one another, meaning
set includes compounds that differ in the property that is comple-
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Fig. 28. Illustration of solute and solvent general adsorption compared to localization. (a) Non-localized retention of chlorobenzene on silica with dichloromethane as mobile
phase; (b) localized retention of phenol, with tetrahydrofuran as mobile phase.
Reprinted with permission from [88].
mentary to the one that is not plotted (i.e., solute HB acidity paired
8.3. NPLC selectivity triangle
with considerations of system HB basicity). The limitation of threeparameters is overcome by creating four different triangles, but the
In addition to the recent uses of triangles in MEKC and RPLC
different plots can lead to different conclusions regarding similar
detailed above, Snyder has recently reviewed solvent selectivity
and different phases. So the need for multiple plots can compli-
and its applications to NPLC [88]. The first part of the review makes
cate the use of the selectivity triangles for selecting orthogonal (or
it clear that the main mechanisms of solute retention on polar sur-
similar replacement) phases.
faces – the work focuses on alumina and silica adsorbents – are (1)
Lastly, as pointed about above, the use of normalized parameters
non-localized adsorption to the surface via displacement of mobile
complicates the chemical interpretation of the parameters. Zhang
phase molecules and (2) localized interactions with the formation
and Carr are careful in their manuscript to avoid labeling the sides
of specific interactions between analytes and the stationary phase.
of the triangles with absolute descriptors as Fu and Khaledi did. In
The former predominate for non-polar solutes and the latter for
fact, they explicitly recognize that the normalization complicates
compounds containing polar function groups. Both types of inter-
interpretations of the derived parameters. This does not preclude
actions are depicted in Fig. 28.
the use of the triangles in a practical sense but does limit the degree
Snyder focuses on the effects of the "B-solvent" – the more polar
to which chemical meaning can be ascribed to conclusions based
solvent in a mixed mobile phase. The A-solvent is typically some-
on them regarding similar and different phases.
thing like n-pentane or cyclohexane. He points out that if a polar,
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
(9) 1-naphthaldehyde; (10) 1,5-dinitronaphthalene; and (11)1-acetonaphthalene.
The first plot in this figure compares the two non-localizing B-
solvents benzene and carbon tetrachloride. The log k values arehighly correlated, meaning that the retention mechanisms withboth phases are essentially identical. When a localizing B-solvent(acetonitrile) is compared to a non-localizing one (benzene), thecorrelation is poor, indicating different interactions are governingretention, creating potential selectivity differences with the differ-ent mobile phases. In these plots, it is clear that polar, localizingsolutes such as 2-methoxynaphthalene, 2-nitronaphthalene, and1-naphthaldehyde are each affected differently by the presence ofacetonitrile in the mobile phase.
To further understand and differentiate the effects of differ-
ent B-solvents, Snyder correlates ı log k values from one �–� plotwith ı log k values from a second �–� plot for the eleven solutesshown in Fig. 29. The squares of the correlation coefficients forthese ı log k vs. ı log k plots are shown in Table 7 (adapted from[89]). To aid in understanding the different results, Snyder uses thesolvatochromic selectivity triangle shown in Fig. 30a to differenti-ate highly basic solvents (top shaded portion of the triangle) fromweakly and non-basic solvents (those outside the top shaded por-tion). The specific B-solvents studied and their relative basicitiesaccording to the definitions used to establish the solvent triangleare shown in Fig. 30b, as are their average r2 values from the ı log kvs. ı log k correlations.
It is clear from these results that those solvents classified as
non- or weakly basic (e.g., nitromethane and acetonitrile) producethe strongest correlations whereas the strongly basic solvents (tri-ethylamine, pyridine, ethyl ether, THF) have the lowest correlationcoefficients. It is also clear from the data in the table that cor-relations between two non-basic solvents (upper left quadrant)produce stronger correlations than those between two basic sol-vents (lower right quadrant). Furthermore, correlations betweena non-basic and basic solvent (upper right quadrant) are weakerthan those between two non-basic solvents (upper left quadrant).
All of this indicates that the basic solvents have some additionalmechanism (or mechanisms) of interacting with solutes and/orthe stationary phase that creates additional likelihood for selec-tivity differences to exist between them. Snyder goes on to offerevidence that part of those selectivity differences relates to theability of those solvents to increase retention for proton-donatingsolutes, with the basic solvents increasing retention more than
Fig. 29. Comparison of solvent-type selectivity for two equal-strength mobile
non-basic solvents. This arises because the basic solvents are con-
phases; (a) non-localizing B-solvents (benzene and carbon tetrachloride), (b) one
centrated on the stationary phase surface. These solvent molecules
localizing B-solvent (acetonitrile). Alumina as adsorbent. See text for solutes and
will preferentially interact with and increase the retention of donor
Reprinted with permission from [88].
The practical upshot to all of these studies is that they provide
guidance for optimizing NPLC separations. As detailed earlier in this
localizing solvent, like tetrahydrofuran (THF) is replaced with a
review, seven ‘training' chromatograms are used in a simplex opti-
less polar one, like CH2Cl2, polar solutes will experience decreased
mization scheme when optimizing three parameters. An isocratic
competition for localized interactions with the surface, leading
optimization scheme related to the use of basic localizing, non-
to preferential retention of polar solutes compared to nonpolar
basic, and non-basic localizing solvents in mobile phases of equal
solutes and hence to a change in the selectivity of the separation.
solvent strength is shown in Fig. 31 (see original publication for
In order to better understand the effects of the nature of the
more details). Chromatograms obtained using some of the ‘training'
B-solvent on selectivity, Snyder first defines a solute-specific
mobile phases are shown in Fig. 32a–c and the optimized chro-
property, ı log k. ı log k values are derived by correlations of log k
matogram is shown in Fig. 32d. The training chromatograms show
values obtained using one mobile phase vs. log k values obtained
multiple overlapping peaks and considerably different selectivi-
using a second mobile phase with a different B-solvent (so-called
ties for some solutes. By combining non-basic, basic, and localizing
�–� plots). Such plots are illustrated in Fig. 29 for eleven solutes.
solvents, a minimum resolution of 1.3 was obtained for all com-
The ı log k parameter is also shown in the figure. The mobile phases
ponents. Comparing chromatograms b and c and the resulting
are adjusted such that their overall solvent strengths, as measured
chromatogram in d shows the profound effect that the addition
by the solvent strength parameter, ε, are comparable. The solutes
of ACN and CH2Cl2 to the mobile phase in (c) has on selectivity for
are (1) 2-methoxynahthalene; (2) 1,7-dimethoxyaniline; (3) 1-
solute pairs 6 + 10, 4 + 11, and 8 + 9 (an unfavorable influence for the
nitronaphthalene; (4) 2-chloroquinoline; (5) 1-methylnaphthoate;
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Table 7
Squares of correlation coefficients for ı log k values from one �–� plot correlated with ı log k values from a second �–� plot based on eleven solutes listed in the text studied
in nine solvents.
Dimethylsulfoxide DMSO
Tetrahydrofuran THF
Reprinted from [89], with permission from Elsevier.
While much of the data upon which Snyder's review rests was
9. Future directions in comparing selectivity
obtained many years ago, the creation of and subsequent develop-ments of the triangle classification system have helped provide a
9.1. A unifying method for comparing chromatographic
more complete understanding of the retention mechanism in NPLC
and continues to serve as a guide for selecting initial chromato-graphic conditions in optimization schemes.
As suggested in Section 1, new methods for determining selec-
tivity differences between chromatographic systems may helpovercome some of the limitations of triangle schemes that wehave described throughout this review. For example, Ishihama andAsakawa constructed vectors in five-dimensional space based onLSER coefficients. They used the angle between vectors to assess thesimilarity of two chromatographic systems [90]. Instead of usingthe angle between two vectors, Abraham and Martins used thedistance between vectors as the metric for comparing two sys-tems [91]. Lázaro et al. also used the distance between vectors,but only after normalizing the vectors to be the same length [92].
Fuguet et al. used principle component analysis of LSER coefficientsfor pseudostationary phases in electrokinetic chromatography,along with radial distribution plots, to assess selectivity differ-ences between the phases [93]. Principle components analysis ofmultiple column parameters such as surface coverage, hydropho-bic selectivity, shape selectivity, hydrogen bonding capacity and
Fig. 30. Solvent-type selectivity as a function of the hydrogen-bond (H-B) basicity of
Fig. 31. Optimization scheme for NPLC using non-localizing, basic-localizing,
the B-solvent. (a) The solvent-selectivity triangle adapted from Ref. [88]; (b) solvent-
and non-basic localizing B-solvents with A-solvents such as pentane, hexane, or
type selectivity as a function of B-solvent H-B basicity.
dichloromethane for ε0 > 0.30.
Reprinted with permission from [88].
Reprinted with permission from [88].
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Fig. 33. Depiction of the selectivity differences between the surface chemistries of
packings (C18, RP18 with an embedded polar group, and phenyl) and the organic
mobile phases methanol and acetonitrile. Light grey values represent ammonium
formate (pH 3). The values on each bar are the measured selectivity differences. Note
that the largest selectivity differences are found along the diagonal lines. Shown
are the selectivity differences between the packing with an embedded polar group
with acetonitrile as the organic modifier and the phenyl and the C18 column with
methanol as the organic modifier.
Reprinted with permission from [96].
largest differences in selectivity (i.e., those most poorly correlated).
Snyder et al. [96] point out that this method requires adjustment ofthe solute retention times such that they span comparable rangesand is only rigorously valid if all of the system variables are indepen-dent. Nevertheless, the presentation of this work was compellingand the approach offers an interesting method for comparing sys-tem selectivities.
We recently presented a 3D visualization cube to visually detect
similarities and differences between separation systems [97]. Ourmethod is based on the correlation of LSER coefficients. Exam-ples of such correlations are shown in Fig. 34, which comparessodium dodecylsulfate with sodium tetradecylsulfate (STS) andwith lithium perfluorooctanesulfonate (LiPFOS). In these analyses,as in Fu and Khaledi's [82] and Zhang and Carr's work [86], all of theLSER coefficients are ratioed to the v-coefficient before the correla-tion is performed. Clearly, SDS and STS correlate strongly, while SDS
Fig. 32. Examples of the application of the scheme in the previous figure for the
selection of an optimum mobile phase. Conditions: 150 × 4.6 Zorbax-SIL column;
mobile phases shown in the figure (50% water-saturated). See original publication
for details of chromatogram recreation based on retention data.
Reprinted with permission from [88].
ion-exchange capacity at pH 2.7 and 7.6 was used by Euerby andPetersson to analyze and easily visualize the similarities and differ-ences between hundreds of RPLC columns [94]. Neue et al. [95,96]recently presented a graphical method for quantifying and visual-izing selectivity differences between chromatographic systems ofvarying pH, eluent type, and stationary phase. Correlations of gra-dient retention times for the same solutes on two different systemsdiffering in one variable (pH, eluent, or stationary phase) are usedto determine s-values, defined as
where r2 is the square of the correlation coefficient. To visualize thedata, prisms are constructed (see Fig. 33) in which the s-value thatcorrelates the two systems are used as tie-lines. In this example, dif-ferences in stationary phase type (C18, RP18 with a polar embeddedgroup, and a phenyl column) are compared, along with differences
Fig. 34. A plot of the correlation of STS and LiPFOS vs. SDS. The axes are defined as
in methanol and acetonitrile as mobile phase modifiers. The pH
the i/v LSER coefficient ratio, where i = a, b, e, or s. STS vs. SDS (�). LiPFOS vs. SDS
of the solution has been held constant in all studies. Systems with
the largest differences in their s-values represent those offering the
Reprinted with permission from [97]. Copyright 2010 American Chemical Society.
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
Table 8
Interpretations of energetic similarity or difference and thus selectivity similarity or different for various combinations of r2, slope, and intercept from correlations of LSER
coefficient ratios from one system vs. those for another system.
Possible results for correlation of s/v, a/v, b/v, and e/v for two systems
v1 = v2
Potentially effective
= 1.00 or /
Potentially effective
= 1.00 or /
= 0.00 or /
Potentially effective
and LiPFOS do not. Given the structural similarity of SDS to STS and
separations that fail on one system may be better on the other. Or,
the dissimilarity of SDS and LiPFOS, these results are not surprising.
the two systems together are candidates to be used as ‘orthogonal'
Such plots are similar to Horvath et al.'s �–� plots [97,98] because
systems in 2D separations. This, of course, assumes that the solute
they fundamentally compare solute retention on one phase to that
properties are such that they take advantage of the differences in
on another. By using LSERs, though, the same compounds need not
the energetics of retention. The elution order of n-alkanes is likely
be run on each system as long as the solutes analyzed on each phase
to be the same in all systems because there is only one dominant
explore a wide range in type and strength of the intermolecular
mode of interaction amongst them. To exploit system differences,
interactions that govern retention. This makes many more system
the solute set must differentially explore the interactions offered
by the systems.
According to Horvath, two systems that yield a �–� plot with
While the paragraph above focuses on finding chemically dif-
a high correlation coefficient and unity slope would be termed
ferent systems, it is also important to point out the utility of the
"homoenergetic" [98]. Linear regressions such as those shown in
approach to finding comparable systems (those with high r2, unity
Fig. 34 yield three statistical metrics: the slope, intercept, and cor-
slopes, and v1 = v2). Such systems can be used as replacements to
relation coefficient of the fit. We have shown [97] that for two
yield comparable separations should such a need arise.
systems whose correlation yields slope = 1.00, intercept = 0.00, and
As noted elsewhere [97] this approach unifies three major con-
r2 = 1.00, and whose v-coefficients are equal, those systems will
cepts in selectivity: (1) the general LSER formalism, or any other
exhibit homoenergetic retention. This implies that the energetics
multi-parameter model of retention such as the Snyder–Dolan
of retention on both phases are identical and thus there is little to
hydrophobic subtraction model, (2) Zhao and Carr's concept that
no difference in their selectivities. In other words, there is no real
the ratios of LSER coefficients, not their absolute magnitudes, are
chance for what Zhao and Carr called ‘effective selectivity' [81].
the important parameters for comparing system selectivity, and
The two systems will yield the same order of elution and thus very
(3) Horvath's �–� plots for classifying systems as homo-, homeo-,
or heteroenergetic.
If the systems exhibit a high correlation and slope = 1.00, but the
v-coefficients for both LSERs are different such that v1/v2 /
the situation would be termed ‘homeoenergetic', indicating a simi-
9.2. Visualizing the results using 3D plots
lar physico-chemical basis for separation but no chances for elutionorder changes [98]. Again Zhao and Carr would say there is no real
The above procedure requires that the LSER coefficient ratios be
chance for effective selectivity differences to exist between the two
analyzed for each pair of systems of interest. For Fu and Khaledi's set
systems [81]. It is possible that the solutes will be spread out on
of 74 MEKC systems, this yields 2701 different comparisons that can
one system more than the other, but the chance for fundamentally
be performed. Performing the correlations is easily automated, but
altering the separation does not exist.
understanding the output could be daunting if one tries to simply
Finally, correlations between systems that yield slopes /
look at the statistical output for this many correlations. A visual-
intercepts /
= 0.00 and/or low correlation coefficients may exhibit
ization method is needed. For that reason, we developed what we
"heteroenergetic retention" [98]. In other words, retention on one
call a system selectivity cube (SSC) – a three-dimensional plot for
phase is not necessarily correlated with retention on the other.
which the axes are the slope, intercept, and correlation coefficient
Potential differences in selectivity exist between the two systems.
for system correlations. Every correlation of LSER coefficient ratios
In fact, elution order changes, and hence ‘effective selectivity dif-
for two systems is then represented as a glyph in three-dimensional
ferences' are possible. When two systems exhibit this kind of
space. The name ‘system selectivity cube' is meant to recognize the
relationship, if the desired separation is not being achieved with
valuable contributions to chromatography arising from Snyder's
one system then switching to the other system could improve the
solvent selectivity triangle. The development and characteristics
of the SSC are detailed elsewhere [97], but an example of the 3D
Thus, correlating the LSER coefficient ratios of one system vs.
plot based on the LSERs gathered by Fu and Khaledi is shown
another and analyzing the slope, intercept, and correlation coef-
ficient can yield information about the similarity or differences
We highlight here a few of the capabilities of this visualization
in selectivity for the two systems. The possible combinations and
their interpretations are shown in Table 8. The key point in Table 8is that retention on the two systems can be compared with three
(1) The light green dot is a marker for the point with slope = 1.00,
parameters: r2, slope, and intercept (a fourth dimension regarding
intercept = 1.00, and r2 = 1.00 (in other words, highly correlated
the relationship between v-coefficients is needed to differentiate
homo- and homeoenergetic retention). Specifically, systems with
(2) The cube can be rotated, shrunk, or enlarged using a mouse to
(1) non-unity slopes, (2) non-zero intercepts, or (3) poor correlation
help highlight certain axes or certain regions of the cube. An
coefficients offer the possibility for ‘effective selectivity differences'
example is shown in Fig. 36.
(i.e., elution order changes, dramatic changes in relative retention,
(3) Different colors represent correlations between systems within
etc.). In other words, their retention mechanisms are different and
a group accordingto the groupings proposed by Fu and Khaledi
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
(A) Display only particular group correlations (e.g., correlations
within group B).
(B) Display (or not) the maximum and minimum values on the
(C) Change the maximum and minimum values for all of the axes.
The large grey ball at one corner indicates the minimum for allthree coordinates.
(D) Display (or not) the light green marker that represents highly
correlated systems.
(E) Display the numeric coordinates of the points shown on the
screen by hitting the ‘output' button. It is important to note herethat not only the slopes and intercepts are provided, but uncer-tainties in these values are also shown. Values of the correlationcoefficients are also shown.
(F) Display only comparisons of interest by selecting individual
systems (e.g., specify system 12 correlated with systems 19,22, 35, and 65).
(G) Add another dimension of data by displaying the v-coefficient
ratio for each comparison. Recall that when the v-coefficients oftwo systems are equal, different energetics exist than when thev-coefficients have different magnitudes. Thus, we have builtin an option to display ‘spikes' on the dots – the ‘spikier' the
Fig. 35. Example of a 3D visualization made by plotting the slope, correlation coef-
dot, the larger the v-coefficient ratio is for the two systems
ficient, and intercept resulting from the correlation of LSER coefficient ratios for two
represented by the glyph.
systems. Each point represents the regression results obtained by correlating two
(H) A free version of the analysis and visualization software is avail-
systems. Data from Fu and Khaledi's MST compilation was used to generate this
plot. See text for other details. The correlation coefficient axis goes from 0.00 (left)to 1.00 (right). The point representing an ideal homeoenergetic relationship has anr2 = 1.00, intercept = 0.00, and slope = 1.00. It is therefore on the rightmost face of the
9.3. Advantages of the system selectivity cube
cube, roughly in the center and indicated with an arrow.
Reprinted with permission from [97]. Copyright 2010 American Chemical Society.
There are some significant benefits to the cube visualization
compared to triangles:
based on their selectivity triangle (blue = group A, red = groupB, green = group C, orange = group D).
• Basing the 3D comparisons on LSERs means that dozens, and
(4) The pink glyphs represent comparisons between systems in
sometimes hundreds of solutes have been used to generate the
different groups.
coefficients. This is likely more reliable than selecting only three
(5) A user interface allows the user to
representative solutes as was done in the early selectivity triangleschemes.
• Additionally, all of the LSER coefficients are simultaneously con-
sidered, unlike triangles which, even if based on LSERs, can onlyconsider three parameters at a time.
• As shown above for the Fu and Khaledi and Zhang and Carr
reports, four separate plots are required to represent all of theparameters when using triangles. With the new approach, a sin-gle plot incorporates all of the data.
• Furthermore, if two systems are the same in three of the four
LSER parameters, they will appear in the same group in one ofthe triangles and potentially in different groups in the other threetriangles. Here, if the fourth parameter is different enough toruin the correlation, the systems will immediately appear to bedifferent. See, for example, the plot for SDS vs. LiPFOS in Fig. 34.
• Unlike �–� plots which require the same solutes to be analyzed
on both columns, with the LSER approach any representative setof solutes can be used to obtain the coefficients upon which themethodology relies.
• Lastly, and perhaps most importantly, this approach can be
applied to any multi-parameter model of retention – not justLSERs. For example, we have started analyzing the large RPLC dataset of Zhang and Carr discussed above which uses the hydropho-bic subtraction model to understand retention.
Fig. 36. Same plot as in the previous figure but rotated to make the correlation
9.4. Disadvantages of the system selectivity cube
coefficient axis more prominent. The default axes values are the high and low valuespresent in the data set but are not shown for clarity. The correlation coefficient axesgoes from 0.00 (left) to 1.00 (right).
Some of the advantages of the new approach are also disad-
Reprinted with permission from [97]. Copyright 2010 American Chemical Society.
vantages, depending on the analyst's goals.Here is a brief (and not
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
necessarily exhaustive) list of some of the important issues as we
set, the two systems could provide nearly identical selectivities
currently see them.
since the solutes cannot take advantage of the difference in thea/v ratios of the two systems. This could be mitigated with proper
• LSER coefficients generally have large relative uncertainties due
weighting schemes, which we are considering. Ideally, the user
to imperfections in the model. Thus, the slopes and intercepts of
would get to input the weighting schemes and in this way get
the correlations may be unreliable as a basis for firmly classify-
to emphasize the solute characteristics they believe to be most
ing similar and dissimilar systems. Nevertheless, we believe the
approach can certainly serve as a helpful guide in distinguishingsystems.
Thus, much work remains to figure out how best to use this
• Another disadvantage is the severe data reduction that is taking
approach and to apply it to other data sets.
place and the chemical information that is lost in the process.
We introduced the SSC and the other methods for analyzing
Hundreds of solutes are sometimes used to generate LSER equa-
selectivity summarized earlier as alternatives and possible com-
tions for a system. The LSERs themselves generally have five
plements to or replacements for selectivity triangles. This brings
or six fitting parameters (therefore 10–12 values for two sys-
us back full circle to the poem in our introduction. At some point,
tems). Thus, in comparing two systems, hundreds of individual
people stopped building pyramids, but as Jennifer Michael Hecht
data points that relate directly to retention and selectivity, and
urges "we must not curse the passage of time."
up to a dozen LSER terms that also relate to solute retention,are getting simplified down to three or four parameters (slope,intercept, correlation coefficient, and v-ratio). These four param-
10. Summary
eters are not immediately related to solute retention. Thus, alot of useful chemical information is not being used explicitly in
Overall, we have traced the development of chromatographic
this model. Such a process has an analogy in assigning semester
selectivity triangle schemes over the past 50 years. Valid criticisms
grades at most U.S. academic institutions. Throughout a semester,
of some of the schemes were considered, and recent applications
students take multiple tests and quizzes, write lab reports, turn
based on new models of retention were highlighted. Finally, newer
in homework, etc. – all of which contain very specific informa-
methods for comparing system selectivity were presented.
tion about student performance. The scores on these individual
Specifically, we started this review by examining the early ori-
pieces get weighted and then averaged together to produce an
gins of triangles as first applied to GC stationary phases. We then
overall semester average. This average is further simplified to
focused largely on Snyder's original SST because it has received the
a single semester grade on a five-grade scale of A through F.
most attention of all of the triangles produced. We discussed the
So a lot of specific information is sacrificed for the simplicity of
use of the SST in combination with simplex experimental designs
obtaining a single letter grade. Likewise, our approach sacrifices
to optimize LC separations. We have also discussed the complica-
specific retention information for the sake of obtaining a simple
tions that the presence of water in RPLC mobile phases causes when
three-parameter comparison of system selectivity.
using selectivity triangles to make accurate predictions of selectiv-
• Another concern that this approach shares with the triangles is
ity. This stems from the effect that water has on the overall polarity
the loss of chemical insight regarding the actual type and strength
of the mobile phase, the specific changes it induces in the organic
of intermolecular interactions governing separations. The glyph
additives, and the specific polar and hydrogen bonding interactions
merely tells the user if two systems are similar or different in
it has with solutes. Because of these effects, even mobile phases of
their energetics of retention but without any details regarding
comparable solvent strength yield different selectivities. Because
the specific blend of interactions. For this, an analysis of the LSERs
the SST is based on pure solvents and does not incorporate the
(or other model) would still need to be done.
effects of water, this generally limits the accuracy of predictions
• As noted above, different amounts of water in RPLC mobile
of RPLC selectivity that are based on the SST.
phases can have dramatically different effects on selectivity, and
With regards to GC, many groups have used the SST approach
the extent of those effects varies with the organic modifier.
to characterize common stationary phase coatings. In the original
As a result, the coefficients of LSERs vary with percent water.
development of the SST, Snyder tried to remove the dispersion
Thus, multiple LSERs would be required to fully characterize one
effects by normalizing partition coefficients of polar solutes to
organic modifier. The proposed selectivity cube could handle this
those of hypothetical n-alkanes of comparable size. Furthermore,
in terms of comparing one modifier at one composition either to
he based the SST apices on test solutes that do not explicitly repre-
the same modifier at a different composition or to a different
sent dispersion interactions. In subsequent studies by other groups,
modifier at the same percent composition. In fact, any number of
changing the test solutes was observed to change the location of the
combinations of modifier/composition could be compared, but
phases within the triangle. The contributions of dispersion interac-
the number of comparisons could be quite large and thus diffi-
tions to selectivity were of specific interest in these studies, as well
cult to fully evaluate. Furthermore, because of the different effects
as some studies involving RPLC.
of water on different solvents, such comparisons will always be
Fifteen years after the publication of Snyder's original SST, sol-
system-specific and will not lead to general classifications regard-
vatochromic parameters were used to show that the original Xe,
ing the organic modifiers.
Xd, and Xn values represented blends of two or more intermolec-
• Somewhat related to this, two systems may be poorly correlated
ular interactions. In subsequent publications, a triangle based on
because of a single parameter. For example, the a/v ratio could be
solvatochromic solvent parameters was presented. This produced
positive for one system and negative for another, with all other
some advantages over the original version in terms of the chem-
ratios generally the same. This is likely to still lead to a poor corre-
ical interpretation of the triangle because of the relative ‘purity'
lation and be interpreted as arising from two dissimilar systems.
of the parameters used to define the apices. Nevertheless, these
Thus, differences in a single parameter may be overemphasized
parameters cannot account for the effects of water on the nature
in this approach (see again the correlation of SDS vs. LiPFOS in
of the solvents. Thus, triangles based on solvatochromic parame-
Fig. 34). This would be particularly important if the user's solute
ters still suffer from the same complications regarding the practical
set does not contain solutes that are hydrogen bond donating (i.e.,
application of triangles to predict RPLC selectivities.
no solutes with significant A values). Our approach would lead
More recently, selectivity triangles have been used in combi-
one to believe that the systems are different, but for such a solute
nation with LSERs to characterize pseudo phases used in MEKC
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
separations. The definition of the parameters, however, com-
[2] R. Shepherd, R. Shepherd, 100 Symbols: What Shapes Mean in Art and Myth,
plicates understanding their chemical meaning and potentially
Thames and Hudson, London, 2002.
[3] L.S. Ettre, LC–GC North Am. 24 (2006) 680.
influences the groupings produced within the triangle. Using simi-
[4] J. Michael Hecht, The Next Ancient World, Tupelo Press, North Adams, 2001.
larly defined parameters, multiple triangles examining hundreds of
[5] I. Brown, Nature 188 (1960) 1021.
commercially available RPLC phases have recently been published.
[6] I. Brown, J. Chromatogr. 10 (1963) 284.
[7] L.R. Snyder, J. Chromatogr. 92 (1974) 223.
These triangles, however, were based on the coefficients produced
[8] L. Rohrschneider, J. Chromatogr. 22 (1966) 6.
by the hydrophobic subtraction model of retention rather than on
[9] W.T. Cooper, P.L. Smith, J. Chromatogr. 355 (1986) 57.
[10] L.R. Snyder, J. Chromatogr. Sci. 16 (1978) 223.
NPLC triangles were recently reviewed by Snyder. These trian-
[11] L.R. Snyder, J.J. Kirkland, Introduction to Modern Liquid Chromatography, 2nd
ed., Wiley–Interscience, New York, 1979.
gles emphasized the importance of solvent interactions with the
[12] W.O. McReyolds, J. Chromatogr. Sci. 8 (1970) 685.
bare stationary phase – either through general non-localized or
[13] W.O. McReynolds, Gas Chromatographic Retention Data, 5th ed., Preston Indus-
specific localized interactions. Furthermore, the influence of sol-
trial, Inc., Niles, IL, 1966.
[14] M.S. Klee, M.A. Kaiser, K.B. Laughlin, J. Chromatogr. 279 (1983) 681.
vent basicity on selectivity was also considered. Because of the
[15] J.P. Teas, J. Paint Technol. 40 (1968) 19.
absence of water and its associated complications, predictions of
[16] C.M. Hansen, J. Paint Technol. 39 (1967) 104.
selectivity based on NPLC triangles schemes are generally much
[17] K. Nicolaus, C. Westphal (Eds.), The Restoration of Paintings, Konemann Ver-
lagsgesellschaft mbH, Cologne, 1999, p. 360.
more accurate than those in RPLC.
[18] J. Wadum, Vermeer Illuminated: A Report on the Restoration of the View of
Lastly, we closed this review by discussing other methods for
Delft and The Girl with a Pearl Earring by Jonannes Vermeer, V+K Publish-
comparing the selectivity of separation systems, focusing on our
ing/Inmerc, Mauritshuis, The Hague, 1994, p. 22.
[19] J.L. Glajch, J.J. Kirkland, K.M. Squire, J.M. Minor, J. Chromatogr. 199 (1980) 57.
system selectivity cube. This work unifies concepts that underpin
[20] S.L. Morgan, S.N. Deming, J. Chromatogr. 112 (1975) 267.
selectivity triangles with theories regarding the homo-, homeo-,
[21] M.W. Watson, P.W. Carr, Anal. Chem. 51 (1979) 1835.
and heteroenergetic nature of retention on two different systems.
[22] R.D. Snee, Chemtech 9 (1979) 702.
[23] S.R. Bakalyar, R. McIlwrick, E. Roggendorf, J. Chromatogr. 142 (1977) 353.
A 3D visualization method for simultaneously viewing hundreds
[24] J.L. Glajch, J.J. Kirkland, Anal. Chem. 55 (1983) 319A.
or thousands of system comparisons was discussed. Potential
[25] J.L. Glajch, J.J. Kirkland, L.R. Snyder, J. Chromatogr. 238 (1982) 269.
advantages and disadvantages of this new approach were briefly
[26] J.J. Kirkland, J.L. Glajch, J. Chromatogr. 255 (1983) 27.
[27] Sz. Nyiredy, B. Meier, C.A.J. Erdelmeier, O. Sticher, J. High Resolut. Chromatogr.
Chromatogr. Commun. 8 (1985) 186.
Given all of the advances that have occurred over the past half
[28] G. D'Agostino, F. Mitchell, L. Castagnetta, M.J. O'Hare, J. Chromatogr. Biomed.
of a century in the development of selectivity triangles, it is natural
Appl. 305 (1984) 13.
to end this review by turning to the future. The theory behind the
[29] G. D'Agostino, L. Castagnetta, F. Mitchell, M.J. O'Hare, J. Chromatogr. Biomed.
Appl. 338 (1985) 1.
triangles seems to be well examined, and the advantages and dis-
[30] W.J. Cheong, P.W. Carr, Anal. Chem. 60 (1988) 820.
advantages of various formulations are well understood. Perhaps it
[31] J.H. Park, M.D. Jang, D.S. Kim, P.W. Carr, J. Chromatogr. 513 (1990) 107.
is time to shift the emphasis from creating more triangle schemes
[32] E.B. Tada, L.P. Novaki, O.A. El Seoud, J. Phys. Org. Chem. 13 (2000) 679.
[33] Y. Marcus, Y. Migron, J. Phys. Chem. 95 (1991) 400.
to rigorously evaluating how well they do the job of characteriz-
[34] A.M. Stalcup, D.E. Martire, S.A. Wise, J. Chromatogr. 442 (1988) 1.
ing selectivity that they are designed to do. We therefore suggest
[35] E.D. Katz, K. Ogan, R.P.W. Scott, J. Chromatogr. 352 (1986) 67.
that it would be helpful to have studies aimed at using some of the
[36] K.L. Rowlen, J.M. Harris, Anal. Chem. 63 (1991) 964.
[37] A. Alvarez-Zepeda, B.N. Barnum, D.E. Martire, Anal. Chem. 64 (1992) 1978.
recent publications involving MEKC and RPLC to show how selectiv-
[38] I. Shulgen, E. Ruckenstein, J. Phys. Chem. B 103 (1999) 872.
ity triangles can be used to guide the selection of pseudo phases or
[39] S.D. West, J. Chromatogr. Sci. 27 (1989) 2.
stationary phases for practical separations. In other words, perhaps
[40] S.D. West, J. Chromatogr. Sci. 25 (1987) 122.
[41] J.J. Lewis, L.B. Rogers, R.E. Pauls, J. Chromatogr. 264 (1983) 339.
we need more examination of how reliably the triangles identify
[42] L.C. Tan, P.W. Carr, J. Chromatogr. A 799 (1998) 1.
different and comparable phases for actual mixtures of interest, not
[43] L.R. Snyder, P.W. Carr, S.C. Rutan, J. Chromatogr. A 656 (1993) 537.
just general test mixtures used to develop LSERs and the hydropho-
[44] J.H. Hildebrand, R.L. Scott, The Solubility of Non-electrolytes, 3rd ed., Dover
bic subtraction model. More specifically, perhaps such schemes can
Publications, New York, 1964.
[45] B.L. Karger, L.R. Snyder, C. Eon, Anal. Chem. 50 (1978) 2126.
be shown to be effective for identifying orthogonal phases that can
[46] E.F. Meyer, R.E. Wagner, J. Phys. Chem. 70 (1966) 3162.
be coupled in 2D RPLC separations. Relatedly, it is equally important
[47] E.F. Meyer, T.A. Renner, K.S. Stec, J. Phys. Chem. 75 (1971) 642.
to be able to identify systems that have comparable selectivities
[48] E.F. Meyer, K.S. Stec, R.D. Hotz, J. Phys. Chem. 77 (1973) 2140.
[49] E.F. Meyer, M.J. Awe, R.E. Wagner, J. Chem. Eng. Data 25 (1980) 371.
so that replacement systems can be readily adopted should some
[50] B.R. Kersten, C.F. Poole, J. Chromatogr. 452 (1988) 191.
commercial phases become unavailable. Such studies, if successful,
[51] C.F. Poole, S.K. Poole, Chem. Rev. 89 (1989) 377.
would then encourage the further development of fundamen-
[52] P. Shah, H. Na, L.B. Rogers, J. Chromatogr. 329 (1985) 5.
[53] T.J. Betts, J. Chromatogr. 354 (1986) 1.
tal triangles schemes for characterizing, selecting, and optimizing
[54] S.T. Preston, J. Chromatogr. Sci. 8 (1970) 18A.
separation systems.
[55] W.T. Cooper, L.Y. Lin, Chromatographia 21 (1986) 335.
[56] R.M. Smith, Anal. Chem. 56 (1984) 256.
[57] S.C. Rutan, P.W. Carr, W.J. Cheong, J.H. Park, L.R. Snyder, J. Chromatogr. 463
[58] M.J. Kamlet, J.L. Abboud, R.W. Taft, J. Am. Chem. Soc. 99 (1977) 6027.
[59] M.J. Kamlet, R.W. Taft, J. Am. Chem. Soc. 98 (1976) 377.
We thank Dr. Lloyd Snyder and Professor Peter Carr for their
[60] R.W. Taft, M.J. Kamlet, J. Am. Chem. Soc. 98 (1976) 2886.
helpful comments and discussions throughout the preparation of
[61] M.J. Kamlet, J.L.M. Abboud, M.H. Abraham, R.W. Taft, J. Org. Chem. 48 (1983)
this manuscript. We also thank the reviewers for their helpful
[62] M.J. Kamlet, T.N. Hall, J. Boykin, R.W. Taft, J. Org. Chem. 44 (1979) 2599.
suggestions and detailed consideration of this review. Acknowl-
[63] R.W. Taft, J.L.M. Abboud, M.J. Kamlet, J. Am. Chem. Soc. 103 (1981) 1080.
edgment is made to the Donors of the American Chemical Society
[64] J.H. Park, A.J. Dallas, P. Chau, P.W. Carr, J. Phys. Org. Chem. 7 (1994) 757.
[65] M.F. Vitha, J. Chem. Educ. 78 (2001) 370.
Petroleum Research Fund for partial support of this research.
[66] E.H. Slaats, W. Markovski, J. Fekete, H. Poppe, J. Chromatogr. 207 (1981) 299.
Support was also provided by the Drake University Science
[67] R.M. McCormick, B.L. Karger, Anal. Chem. 52 (1980) 2249.
Collaborative Institute (DUSCI).
[68] R.M. McCormick, B.L. Karger, J. Chromatogr. 199 (1980) 259.
[69] C.R. Yonker, T.A. Zweir, M.F. Burke, J. Chromatogr. 241 (1982) 257.
[70] C.R. Yonker, T.A. Zwier, M.F. Burke, J. Chromatogr. 241 (1982) 269.
[71] R.P.W. Scott, P. Kucera, J. Chromatogr. 142 (1977) 213.
[72] C.S. Koch, F. Köster, G.H. Findenegg, J. Chromatogr. 406 (1987) 257.
[73] A. Tilly-Melin, Y. Askemark, K.G. Wahlund, G. Schill, Anal. Chem. 51 (1979) 976.
[1] J. Tresider (Ed.), The Complete Dictionary of Symbols, Chronicle Books, San
[74] D.E. Martire, R.E. Boehm, J. Phys. Chem. 87 (1983) 1045.
Francisco, 2004.
[75] M.R. Böhmer, L.K. Koopal, R. Tijssen, J. Phys. Chem. 95 (1991) 6285.
Author's personal copy
A.R. Johnson, M.F. Vitha / J. Chromatogr. A 1218 (2011) 556–586
[76] J. Li, Y. Zhang, P.W. Carr, Anal. Chem. 64 (1992) 210.
[88] L.R. Snyder, J. Planar Chromatogr. 21 (2008) 315.
[77] J. Li, Y. Zhang, A.J. Dallas, P.W. Carr, J. Chromatogr. 550 (1991) 101.
[89] L.R. Snyder, J.L. Glajch, J.J. Kirkland, J. Chromatogr. 218 (1981) 299.
[78] J. Li, Y. Zhang, H. Ouyang, P.W. Carr, J. Am. Chem. Soc. 114 (1992) 9813.
[90] Y. Ishihama, N. Asakawa, J. Pharm. Sci. 88 (1999) 1305.
[79] B.R. Kersten, S.F. Poole, C.F. Poole, J. Chromatogr. 468 (1989) 235.
[91] M.H. Abraham, F. Martins, J. Pharm. Sci. 93 (2004) 1508.
[80] J. Li, A.J. Dallas, P.W. Carr, J. Chromatogr. 517 (1990) 103.
[92] E. Lázaro, C. Ràfols, M.H. Abraham, M. Rosés, J. Med. Chem. 49 (2006) 4861.
[81] J. Zhao, P.W. Carr, Anal. Chem. 71 (1999) 2623.
[93] E. Fuguet, C. Ràfols, E. Bosch, M.H. Abraham, M. Rosés, Electrophoresis 27 (2006)
[82] C. Fu, M.G. Khaledi, J. Chromatogr. A 1216 (2009) 1891.
[83] C. Fu, M.G. Khaledi, J. Chromatogr. A 1216 (2009) 1901.
[94] M.R. Euerby, P. Petersson, J. Chromatogr. A 994 (2003) 13.
[84] M.H. Abraham, A. Ibrahim, A.M. Zissimos, J. Chromatogr. A 1037 (2004)
[95] U. Neue, K.J. Fountain, P.C. Iraneta, B.A. Alden, K. Wyndham, Pittcon 2010,
Orlando, FL, paper no. 2890-2, March 4, 2010.
[85] M.F. Vitha, P.W. Carr, J. Chromatogr. A 1126 (2006) 143.
[96] L.R. Snyder, J.W. Dolan, P.W. Carr, R.E. Majors, LC–GC 28 (2010) 418.
[86] Y. Zhang, P.W. Carr, J. Chromatogr. A 1216 (2009) 6685.
[97] A.R. Johnson, M.F. Vitha, T. Urness, T. Marrinan, Anal. Chem. 82 (2010) 6251.
[87] L.R. Snyder, J.W. Dolan, P.W. Carr, J. Chromatogr. A 1060 (2004) 77.
[98] W. Melander, J. Stoveken, C. Horvath, J. Chromatogr. 199 (1980) 35.
Source: http://craiguniversity.tk/media/departmentsoffices/dusci/pdfdocuments/Chromatography-selectivity-triangles.pdf
Tai Chi and Postural Stability in Patients with Parkinson's Disease Fuzhong Li, Ph.D., Peter Harmer, Ph.D., M.P.H., Kathleen Fitzgerald, M.D., Elizabeth Eckstrom, M.D., M.P.H., Ronald Stock, M.D., Johnny Galver, P.T., Gianni Maddalozzo, Ph.D., and Sara S. Batya, M.D. BACKGROUNDPatients with Parkinson's disease have substantially impaired balance, leading to di- From the Oregon Research Institute (F.L.),
[Boletín de Infraestructuras de Burgos] [Cuarto trimestre 2011] Boletín de Infraestructuras de Burgos [ASOCIACIÓN PLAN ESTRATÉGICO DE BURGOS] [En este número] Ha pasado un año desde la publicación del último boletín. En este tiempo, la situación económica del país no ha permitido grandes novedades en el estado de la construcción de nuevas infraestructuras, salvo en la apertura

