پرستاری خون
نوخ یراتسرپ
متفه هسلج
THE PATIENT WITH SICKLE CELL CRISIS
NURSING DIAGNOSES:
• • Acute pain related to tissue hypoxia due to agglutination of
sickled cells within blood vessels
• • Risk for infection• • Risk for powerlessness related to illness-induced
• • Deficient knowledge regarding sickle crisis prevention.
COLLABORATIVE PROBLEMS/
POTENTIAL COMPLICATIONS
• • Hypoxia, ischemia, infection, and poor wound healing leading to
skin breakdown and ulcers
• • Dehydration• • Cerebrovascular accident (CVA, brain attack, stroke)• • Anemia• • Renal dysfunction• • Heart failure, pulmonary hypertension, and acute chest syndrome• • Impotence• • Substance abuse related to poorly managed chronic pain
Planning and Goals
• The major goals for the patient are relief of pain,
decreased incidence of crisis, enhanced sense of self-esteem and power, and absence of complications.
Nursing Interventions
•
MANAGING PAIN:
• Acute pain during a sickle cell crisis can be severe and unpredictable.
• The patient's subjective description and rating of pain on a pain scale must
guide the use of analgesics, which are valuable in controlling the acute pain
of a sickle crisis.
• Any joint that is acutely swollen should be supported and elevated until
the swelling diminishes.
• Relaxation techniques, breathing exercises,and distraction are helpful for
some patients.
• Physical therapy, whirlpool baths, and transcutaneous nerve stimulation
are examples of such modalities.
PREVENTING AND MANAGING INFECTION
• Nursing care focuses on monitoring the patient for signs and
symptoms of infection.
• Prescribed antibiotics should be initiated promptly, and the patient
should be assessed for signs of dehydration.
• If the patient is to take prescribed oral antibiotics at home, he or she
must understand the need to complete the entire course of antibiotic therapy and must be able to identify a feasible administration schedule.
PROMOTING COPING SKILLS
• This illness, because of its acute exacerbations that often
result in chronic health problems, frequently leaves the patient feeling powerless and with decreased self-esteem.
• These feelings can be exacerbated by inadequate pain
• The patient's ability to use normal coping resources of
physical strength, psychological stamina, and positive self-esteem is dramatically diminished.
• Enhancing pain management can be extremely useful in
establishing a therapeutic relationship based on mutual trust.
• Nursing care that focuses on the patient's strengths rather
than deficits can enhance effective coping skills.
• Providing the patient with opportunities to make decisions
about daily care may increase the patient's feelings of control.
MINIMIZING DEFICIENT KNOWLEDGE
• Patients with sickle cell anemia benefit from understanding what
situations can precipitate a sickle cell crisis and the steps they can take to prevent or diminish such crises.
• Keeping warm and maintaining adequate hydration can be very
effective in diminishing the occurrence and severity of attacks.
• Avoiding stressful situations is more challenging.
• Group education may be more effective if it is carried out by
members of the community who are from the same ethnic group as those with the disease.
MONITORING AND MANAGING
POTENTIAL COMPLICATIONS
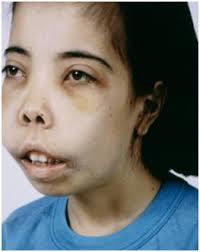
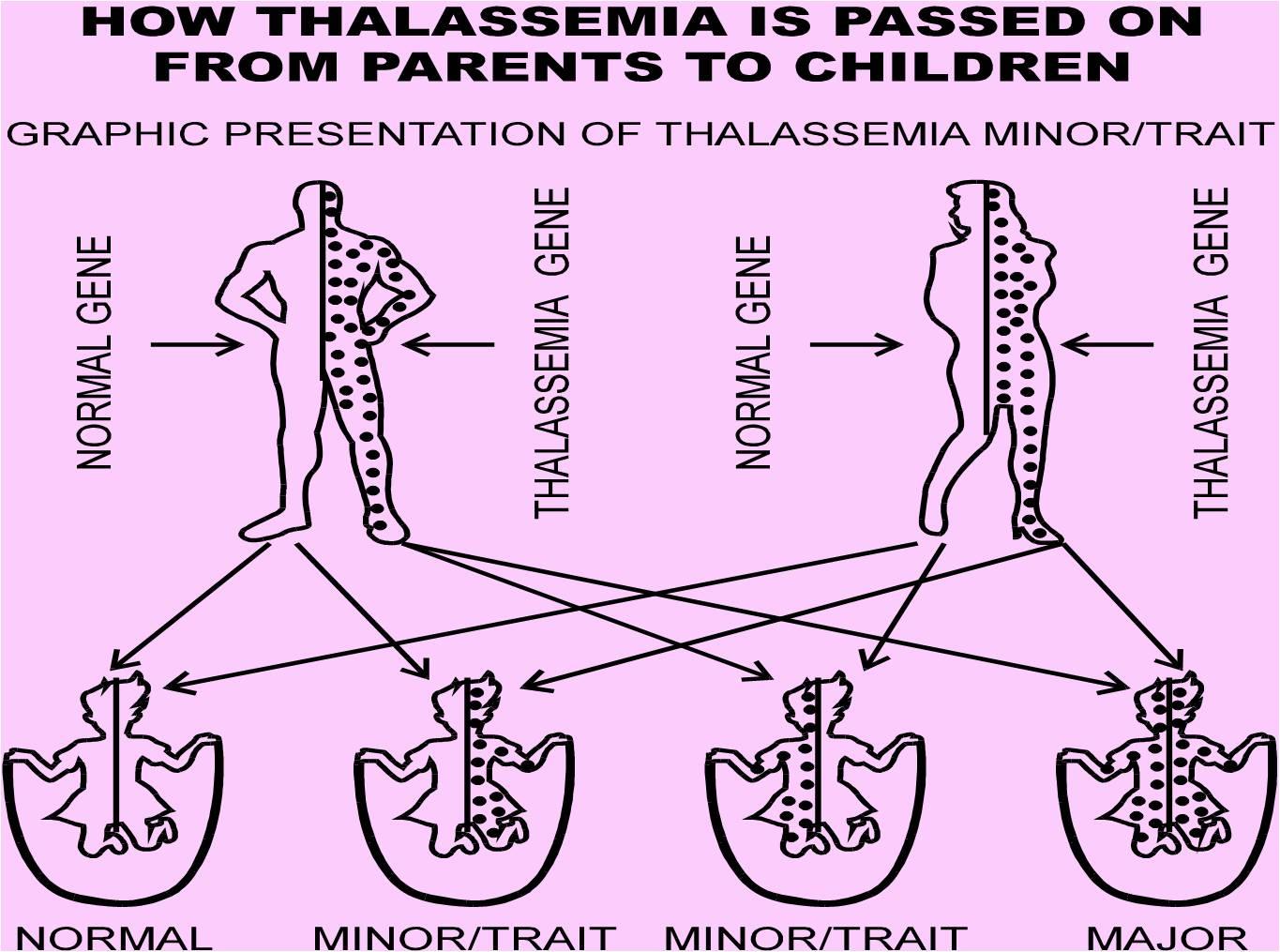
• Causes of Beta
Thalassaemia Beta
thalassemias are caused
by mutations on
chromosome 11.
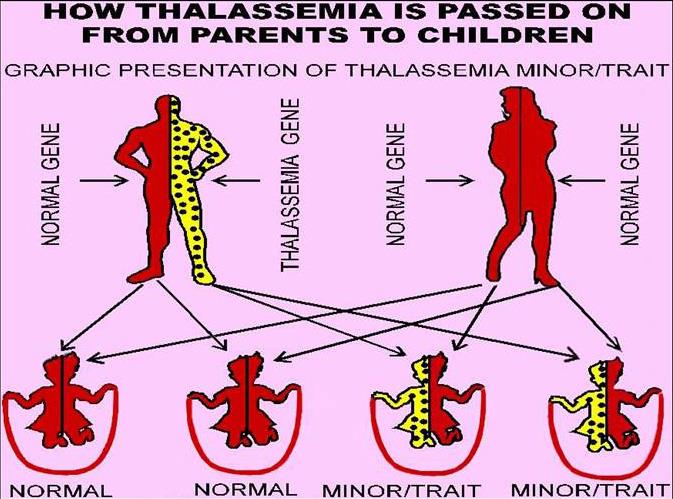
• Thalassemia thalassaemia), also called
• The thalassemias are a group of hereditary disorders associated with
defective hemoglobin-chain synthesis.
• These anemias occur worldwide, but the highest prevalence is found
in people of Mediterranean, African, and Southeast Asian ancestry .
• Thalassemias are characterized by hypochromia (an abnormal
decrease in the hemoglobin content of RBCs), extreme microcytosis
(smaller-than-normal RBCs), destruction of blood elements
(hemolysis), and variable degrees of anemia.
• In thalassemia, the production of one or more globulin
chains within the hemoglobin molecule is reduced.
• When this occurs,the imbalance in the configuration of the
hemoglobin causes it to precipitate in the erythroidprecursors or the RBCs themselves.
• This increases the rigidity of the RBCs and thus the
premature destruction of these cells.
• The thalassemias are classified into two major groups
according to the globin chain diminished: alpha and beta.
• The alphathalassemias occur mainly in people from Asia and
the Middle East; the beta-thalassemias are most prevalent in Mediterranean populations but also occur in people from the Middle East or Asia.
• The alpha-thalassemias are milder than the beta forms and
often occur without symptoms. The RBCs are extremely microcytic,but the anemia, if present, is mild.
• Patients with mild forms have a microcytosis and mild anemia.
• If left untreated,severe beta-thalassemia (thalassemia major, or
Cooley's anemia) can be fatal within the first few years of life.
• If it is treated with regular transfusion of RBCs, patients may survive
into their 20s and 30s.
• Etymology references
• Cooley's anemia
• First use: about 1935
• Origin: Thomas B. Cooley died 1945 American pediatrician
Thalassemia Major
• Thalassemia major (Cooley's anemia) is characterized by severe anemia,
marked hemolysis, and ineffective erythropoiesis (production of RBCs).
• With early regular transfusion therapy, growth and development through
childhood are facilitated.
• Organ dysfunction due to iron overload results from the excessive
amounts of iron obtained through the RBC transfusions.
• Regular chelation therapy (eg, via subcutaneous deferoxamine) has
reduced the complications of iron overload and prolonged the life of these
• This disease is potentially curable by BMT if the procedure can be
performed before damage to the liver occurs (ie, during childhood).
DEHYDROGENASE DEFICIENCY
Vicia faba, also known as the broad bean,
fava bean, faba bean, field bean,
bell bean, or tic bean,
is a species of flowering plant in the and family
It is native to –] southwest and s, and
extensively cultivated elsewhere.[
Vicia faba var. equina Pers. – horse bean has
been previously recognized.[
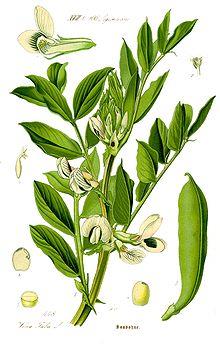
• Glucose-6-phosphate dehydrogenase deficiency (G6PD
deficiency) also known as favism (after the ) is an
genetic condition that predisposes to
(spontaneous destruction of resultanin response to a number of triggers, such
as certain foods, illness, or medication.
• It is particularly common in people of Mediterranean and
African origin.
• The abnormality in this disorder is in the G-6-PD gene; this gene produces
an enzyme within the RBC that is essential for membrane stability.
• A few patients have inherited an enzyme so defective that they have a
chronic hemolytic anemia; however, the most common type of defect
results in hemolysis only when the RBCs are stressed by certain situations,
such as fever or the use of certain medications.
• The disorder came to the attention of researchers during World War II,
when some soldiers developed hemolysis while taking primaquine, an
antimalarial agent.
• African Americans and people of Greek or Italian origin are those primarily
affected by this disorder.
• All types of G-6-PD deficiency are inherited as X-linked
defects; therefore, many more men are at risk than women.
• In the United States, about 12% of African American males
are affected.
• Medications that have hemolytic effects for people with G-6-
PD deficiency are oxidant drugs:
• These medications include antimalarial agents (eg, chloroquine
,sulfonamides (eg,trimethoprim and sulfamethoxazole) ,nitrofurantoin ,common coal tar analgesics (including aspirin in high doses), thiazide diuretics (eg, hydrochlorothiazide ) ,oral hypoglycemic agents (eg,glyburide ,metformin ,chloramphenicol ,and vitamin K (phytonadione).
• In affected people, a severe hemolytic episode can result from
ingestion of fava beans.
Clinical Manifestations
• Patients are asymptomatic and have normal hemoglobin
levels and reticulocyte counts most of the time.
• However, several days after exposure to an offending
medication, they may develop pallor,jaundice, and hemoglobinuria (hemoglobin in the urine).
• The reticulocyte count rises, and symptoms of hemolysis
• Special stains of the peripheral blood may then
disclose Heinz bodies (degraded hemoglobin) within the RBCs.
• Hemolysis is often mild and self-limited. • However, in the more severe Mediterranean type of
G-6-PD deficiency, spontaneous recovery may not occur and transfusions may be necessary
Assessment and Diagnostic Findings
• The diagnosis is made by a screening test or by a
quantitative assayof G-6-PD.
Medical Management
• The treatment is to stop the offending medication. • Transfusion is necessary only in the severe hemolytic
state, which is more commonly seen in the Mediterranean variety of G-6-PD deficiency.
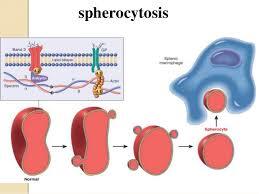
HEREDITARY SPHEROCYTOSIS
• Hereditary spherocytosis is a
relatively common (1 in 5,000 people)hemolytic anemia characterized by an abnormal permeability of the RBC membrane; this permits the cells to change into a spherical shape.
• These RBCs are destroyed prematurely in the spleen. • The severity of this hemolytic anemia varies; jaundice can be
intermittent,and splenomegaly (enlarged spleen) also can occur.
• Surgical removal of the spleen is the principal treatment for
this disorder.
IMMUNE HEMOLYTIC ANEMIA
• Hemolytic anemias can
result from exposure of the RBC to antibodies.
• Alloantibodies (ie, antibodies against the host, or "self")
result from the immunization of an individual with foreign antigens (eg, the immunization of an Rh-negative person with Rh-positive blood).
• Alloantibodies tend to be large (IgM type) and cause
immediate destruction of the sensitized RBCs, either within the blood vessel (intravascular hemolysis) or within the liver.
• The most common type of alloimmune hemolytic anemia in
adults results from a hemolytic transfusion reaction.
• Autoantibodies are developed by an individual for varying reasons.
• In many instances, the person's immune system is dysfunctional,so
that it falsely recognizes its own RBCs as foreign and produces antibodies against them.
• This mechanism is seen in people with chronic lymphocytic leukemia
• Another mechanism is a deficiency in suppressor lymphocytes, which
normally prevent antibody formation against a person's own antigens.
• Autoantibodies tend to be of the IgG type.
• The RBCs are sequestered in the spleen and destroyed by the
macrophages outside the blood vessel (extravascular hemolysis).
• Autoimmune hemolytic anemias can be classified based on the body
temperature involved when the antibodies react with the RBC antigen.
• Warm-body antibodies bind to RBCs most actively in warm
conditions (37°C); cold-body antibodies react in cold (0°C).
• Most autoimmune hemolytic anemias are the warm-body type.
• Autoimmune hemolytic anemia is associated with other disorders in
most cases (eg, medication exposure, lymphoma, CLL,othermalignancy, collagen vascular disease, autoimmune disease,infection).
• In idiopathic autoimmune hemolytic states, the reason why the
immune system produces the antibodies is not known.
• All ages and genders are equally vulnerable to this form, whereas the
incidence of secondary forms is greater in people older than 45 years of age and in females.
Clinical Manifestations
• The hemolysis may be very mild, so that the patient's marrow
compensates adequately and the patient is asymptomatic.
• At the other extreme, the hemolysis can be so severe that the anemia
• Most patients complain of fatigue and dizziness.
• Splenomegaly is the most common physical finding,occurring in more
than 80% of patients; hepatomegaly, lymphadenopathy,and jaundice are also common.
Assessment and Diagnostic Findings
• The laboratory tests show a low hemoglobin level and
hematocrit, most often with an accompanying increase in the reticulocyte count.
• RBCs appear abnormal; spherocytes are common. • The serum bilirubin level is elevated.
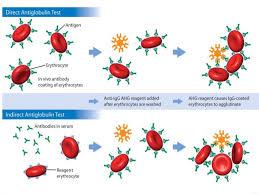
• The Coombs test (also referred to as the direct antiglobulin
test [DAT]), which detects antibodies on the surface of RBCs, shows a positive result.
Medical Management
• Any possibly offending medication should be immediately
• The treatment consists of high doses of corticosteroids (1 mg/kg per
day) until hemolysis decreases.
• Corticosteroids decrease the macrophage's ability to clear the
• If the hemoglobin level returns toward normal, usually after several
weeks, the corticosteroid dose can be lowered or, in some cases, tapered and discontinued.
• However, corticosteroids rarely produce a lasting remission.
• In severe cases, blood transfusions may be required.
• Because the antibody may react with all possible donor
cells, careful blood typing is necessary, and the transfusion should be administered slowly and cautiously.
• Splenectomy (removal of the spleen) removes the major site of RBC
destruction; therefore, splenectomy may be performed if corticosteroids do not produce a remission.
• If neither corticosteroid therapy nor splenectomy is successful,
immunosuppressive agents may be administered.
• The two immunosuppressive agents most frequently used are
cyclophosphamide (eg, Cytoxan), which has a more rapid effect but more toxicity, or azathioprine (Imuran),which has a less rapid effect but less toxicity.
• The synthetic androgen danazol (Cyclomen, Danocrine) can be useful
in some patients, particularly in combination with corticosteroids.
• The mechanism for this success is unclear.
• If corticosteroids or immunosuppressive agents are used, the taper
must be very gradual to prevent a rebound "hyperimmune" response and exacerbation of the hemolysis.
• Immunoglobulin administration is effective in about one third of
patients, but the effect is transient and the medication is expensive.
• Transfusions may be necessary if the anemia is severe; it may be
extremely difficult to cross-match samples of available units of RBCs with that of the patient.
• For patients with cold-antibody hemolytic anemia, treatment may not
be required, other than to advise the patient to keep warm;relocationto a warm climate may be necessary.
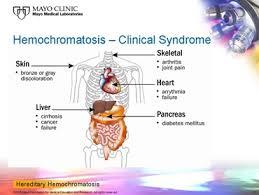
• a genetic condition in which iron
is abnormally(excessively) absorbed from the gastrointestinal tract.
• The excessive iron is deposited
in various organs, particularly the liver, myocardium, testes, thyroid, and pancreas.
• Eventually, the affected organs
• The actual incidence of hemochromatosis is unknown; however,
hereditary hemochromatosis is diagnosed in 0.5% of the population in the United States (ie,1 million people).
• Recent data suggest that this defect may be a common cause of
diabetes (Schechter, et al., 2000).
• Because of to their natural loss of iron through menses, women are
less affected than men.
• Because the accumulation of iron in body organs occurs
gradually,there often is no evidence of tissue injury until middle age.
• Symptoms of weakness, lethargy, arthralgia, weight loss, and loss of
libido are common.
• The skin may be hyperpigmented with melanin deposits (occasionally
hemosiderin, an iron-containing pigment) and appears bronze in
color.
• Cardiac dysrhythmias and cardiomyopathy can occur.
• Endocrine dysfunction is manifested as hypothyroidism, diabetes
mellitus,and hypogonadism (testicular atrophy, diminished libido,andimpotence).
• The most useful laboratory findings are an elevated serum iron level
and high transferrin saturation (more than 60% in men, more than 50% in women).
• The definitive diagnostic test is a liver biopsy.
• Recently, a mutation in the HFE gene has been shown to occur in
most patients with hereditary hemochromatosis (Gochee & Powell, 2001).
• Patients who are homozygous for the gene are at high risk for
development of the disorder.
Medical Management
• Therapy involves the removal of excess iron via therapeutic phlebotomy .
• Each unit of blood removed results in a decrease of 200–250 mg of iron. • The objective typically is to reduce the serum ferritin to less than 50 μg/L
and the transferrin saturation to 35% or less.
• a frequent phlebotomy schedule is required (1 to 2 units weekly), with a
gradual reduction in frequency of phlebotomies over a 1- to 3-year period.
• After 1 to 3 years, the frequency of phlebotomy can be reduced to 1 unit
of blood every several months to prevent reaccumulation of iron deposits.
• Removal of excess iron appears to diminish the severity of diabetes and
skin hyperpigmentation; cardiac function also tends to improve.
The Polycythemias
• Polycythemia refers to an increased volume of RBCs.
• It is a term used when the hematocrit is elevated (to more than 55%
in males, more than 50% in females).
• Dehydration (decreased volume of plasma) can cause an elevated
hematocrit, but not typically to the level to be considered polycythemia.
• Polycythemia is classified as either primary or secondary.
Source: http://www.iausdj.ac.ir/ostad/DocLib8/%D9%86%DB%8C%D9%85%D8%B3%D8%A7%D9%84%20%D8%A7%D9%88%D9%84%2095-94/%D9%BE%D8%B1%D8%B3%D8%AA%D8%A7%D8%B1%DB%8C%20%D8%AE%D9%88%D9%86/%D9%BE%D8%B1%D8%B3%D8%AA%D8%A7%D8%B1%DB%8C%20%D8%AE%D9%88%D9%86%20%D8%AC%D9%84%D8%B3%D9%877.pdf
Good Business…Better Health A comprehensive guide for smoke-free workplaces Acknowledgements: We thank our community partners, fel ow health units and internal departments for their insight and Adapted from the fol owing: Smoking Cessation in the Workplace: A Guide to helping your employees quit smoking. Health Canada, 2005 Towards a Healthier Workplace A Guide Book on Tobacco Control Policies. Health Canada, 2003 Employers' smoking cessation guide: Practical approaches to a costly workplace problem. Second Edition, Date unknown Updated 2015
Lithium-ion batteries – The bubble bursts Stuttgart, October 2012 Consolidation in the lithium-ion battery (LiB) market is inevitable – Stakeholders need to revise their strategies The large-format lithium-ion cell market will face overcapacity and price wars: - Demand is lower than expected - A lot of capacity has been built up – but new equipment to be installed will be

