Ejuh_62
The African Review of Physics (2013)
8:0062 463
Linear and Non Linear Optical Effects of Pyrimethamine and Sulfadoxine:
Ab-initio and Density Functional Study
Geh Wilson Ejuh1, 2,* and Ndjaka Jean Marie1
1University of Dschang, IUT Bandjoun, Department of General and Scientific Studies, Bandjoun, Cameroon
2Université de Yaoundé I, Faculté des Sciences, Département de Physique, Yaounde, Cameroun
The molecular structures of pyrimethamine and sulfadoxine have been explored for non linear optical effects. The ab-
initio Hartee Fock calculations and Density Functional Theory with B3LYP method have been carried out employing 6-311++G** basis set. The dipole moments (µ), polarizability (α), and first hyperpolarizability (βmol) in gas phase and in solvated medium (water and ethanol) are calculated using the same level of theory. The variation of the C-N bond lengths suggests that there is an extended pi-electron delocalization over the pyrimidine moiety that is responsible for non linearity of these molecules. Equally, the large values of βmol for these molecules suggest potential applications of these molecular systems in the development of non linear materials. The large βmol values, which is a measure of the non linear optical activity of the molecular system, is associated with the intermolecular charge transfer resulting from an electron cloud movement through the pi-electron delocalization. Hence theoretical determination of βmol is quite useful, both in the understanding of relationship between the molecular structure and non linear optical properties.
1. Introduction
molecular systems have been studied for predicting nonlinearity [12]. Organic compounds with
Molecular materials with non linear optical (NLO)
electron donating group on one side of the
properties are currently attracting considerable
molecule and electron accepting group on other
attention because of their potentials applications in
side have been studied by experimental and
optoelectronics devices used in telecommunication,
theoretical scientists for NLO properties [13].
information storage, optical switching, signal
Designs of these organic NLO active materials are
processing, and terahertz wave generation [1,2,3,4].
based on the approach of charge transfer due to π-
Non linear optics is one of the few research
electron cloud movement from donor to acceptor
frontiers where tremendous interest arises not only
groups on either side of these π-conjugated
from the desire to understand new physical
systems. This in turn affects the value of
phenomena but also from the potential of
polarizability and hyper-polarizability [7]. It has
technology applications [5].
been known from recent studies that molecular
Organic molecules with delocalized π-electrons
systems, based on electron donor and electron
showing large values of non linear optical
acceptor units connected through pi electron
parameters are gaining interest among researcher
moieties, show many interesting non-linear optical
because of their potential applications in the field
characteristics and have higher value of second
of optoelectronics, such as optical communication,
order NLO properties [14,15,16,17]. The types of
optical computing, optical switching, and image
pi-bridges studied so far for developing efficient
processing [6,7,8,9,10]. The electro-optic effect
NLO materials and molecules include donor-
arises in photorefractive materials (optoelectronic
acceptor acetylenes [18], azo complexes [19],
materials) in two forms; linear electro-optic (LEO)
aromatic ring [14] and hetero-aromatic rings [20].
effect or Pockels effect and quadratic electro-optic
A good deal of work has been done for
(QEO) effect or Kerr effect. Organic molecules are
good materials for nonlinear devices as they are
conjugated polymers consisting of six member and
chemically flexible and show a large and fast non
five-member aromatic units, such as benzene,
linear optical response [11] as a result of which
pyridine, Thiophene, pyrrole, N,N- dimethyl-5-
their optical and electronic properties can be
studied. A number of organic and organometallic
2-amine 5-nitro-2-phenylpyridine, and others
[20,21]. Their derivatives are also considered to be
*
[email protected]
promising materials for electronic and nonlinear
The African Review of Physics (2013)
8:0062 464
optical technology. So far many studies have also
initio and DFT calculations have been carried out
been carried out to investigate the nonlinear
with 6-311++G** basis set.
behavior of two π-conjugated rings attached with a single bond such as biphenyls [20] and
2. Computational Methodology
phenylpyridine [22]. However, pyrimethamine and
All calculations of polarizability and first static
Sulfadoxine can further be investigated as a bridge
hyper-polarizability
for the search of better NLO materials and for an
Sulfadoxine molecules were performed using
understanding of the mechanism at atomic level.
Gaussian 03W [28]. Initial geometry optimizations
Theoretical calculations are quite useful both in
were performed using the ab-initio RHF method
understanding the relationship between molecular
with 3-21G basis set. Subsequently, its results were
structures and nonlinear optical properties, and also
utilized to 6-31G basis set and final calculations
provide guidelines to experimentalists for the
were carried out with double polarized triple zeta
design and synthesis of new organic NLO
split valence 6-311++G** basis set. These
materials. A large number of semi-empirical and ab
structures were refined further using Density
initio calculations have been reported on molecular
Functional Theory, which is a cost effective
hyperpolarizability (β), which is one of the key
method for inclusion of electron correlations with
parameter in the investigation of second order NLO
the three-parameter density functional generally
materials [23,24,25]. Experimental measurements
known as Becke3LYP (B3LYP). This includes
and theoretical calculations on molecular hyper-
Becke's gradient exchange corrections [29], the
polarizability have become one of the key factors
Lee, Yang and Parr correlation functional [30] and
in the determination of second-order NLO material
the Vosko, Wilk and Nusair correlation functional
design. Though, there is an effect of the basis set
[31] with a 6-311++G** basis set. As the first step,
to determine the value of first molecular hyper-
geometry optimizations were carried out and then
polarizability and electronic properties of the
IR and Raman frequencies were calculated using
molecule. The effect is not very high but the
the Hessian, which is the matrix of second
electronic properties and the hyper-polarizability
derivatives of the energy with respect to the
values have been reported to slightly increased on
geometry. We have chosen 6-311++G** basis set,
taking up higher basis sets like the 6-31G and 6-
which has been supplemented by including diffuse
311++G** [18,26]. For this reason we have
and polarization functions in order to ensure proper
decided to carry out our computation using higher
basis set. Equally, as documented in literature,
polarizabilities. This basis set is an adequate basis
accurate estimate of polarizability and hyper-
set and allows for physically correct polarization of
polarizability need polarized and diffused basis sets
the molecule in the presence of an electric field.
as well as introduction of electron correlation
contributions [27].
properties requires the use of extended basis sets
The aim of the present work is to predict NLO
and a high level of theory [22], hence the used of
properties of molecules Pyrimethamine and
B3LYP in our calculations.
sulfadoxine. The idea is to study the charge transfer
The values of mean polarizability (α) of
effect from donor to acceptor through Pyrimidine
molecular systems and the isotropy (γ ) reported in
moiety and determine theoretically the hyper-polarizability of these molecules. This is because
the present work were calculated by using the
following equations
polarizability is quite useful to understand both the
relationship between their molecular structure and
their NLO properties. In calculating the non linear
behavior for the designed molecular systems, ab-
−
yy )2 + (
yy −
zz )
2 + (
zz −
xx )
+ 6
xy +
yz +
The total hyper-polarizability is defined as
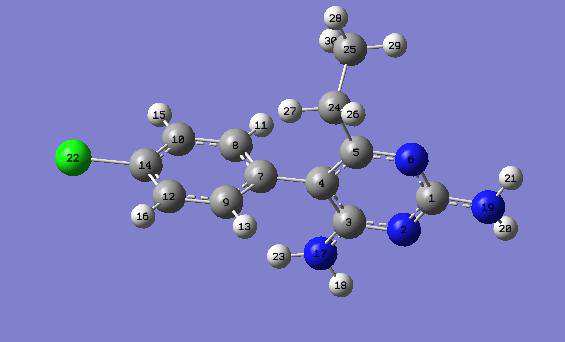
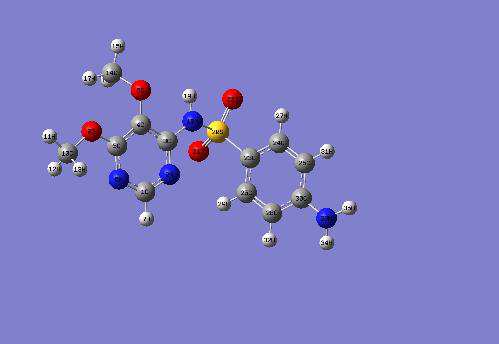
The African Review of Physics (2013) 8:0062 465
The magnitude of βmole is calculated from the computed components β using following equations
( ikk kik kki)
3 i,k
k = x, y, z; i = x, y, z (4)
Fig.2: Optimized structure of Sulfadoxine [26,33].
([ xxx xyy xzz )2
3.1. Optimized geometrical properties of
The geometrical parameters of a Pyrimethamine
molecule in gas phase, water and ethanol are listed
in Tables 1a and 1b. The calculated bond lengths at
3. Molecular Structure and Geometrical
RHF/6-311++G** level are slightly smaller
Properties
(ranging from 0.01Å to 0.04Å) than their corresponding values obtained at the B3LYP/6-
The molecular structure of Pyrimethamine is
311++G** level (Table 1a). It seems that inclusion
shown in Fig. 1 and that of Sulfadoxine is shown in
of electrons correlation expand the molecules.
Fig. 2. The geometric optimization of any system
Once dissolved (in water and ethanol), the bond
gives the ground state geometry of that system. The
lengths remain unaffected at RHF/6-311++G**
total ground state energy of a system is obtained as
level while at B3LYP/6-311++G** level, the bond
a function of the coordinates of the nuclei from
lengths vary slightly except for the following N2-
Born-Oppenheimer (BO) approximation. The
C3, C5-N6, C5-C4, C7-C8, C7-C9, C9-C12, C12-
ground state geometry corresponds to the minimum
C14, C8-H11, C9-H13, C10-H15,C12-H16, C24-
total ground state energy whereas a first order
H27, C25-H28, C25-H29 and C25-H30, all of
saddle point on the BO surface gives the transition
which remain unaffected. The bond lengths (Table
state geometry. Molecular geometries were fully
1a) at the RHF/6-311++G** and at the B3LYP/6-
optimized using Berny's optimization algorithm in
311++G** are approximately equal to the
Gaussian 03, as well as redundant internal
experimental results [34,35,36]. The B3LYP/6-
311++G** results are in better accord with the
experimental results and with the theoretical results given by [37]. For example in the Pyrimidine ring, the C5-N6 and C1-N6 bond lengths are exactly equal to those given by [37] and are 0.017Å and 0.013 Å smaller than the C-N experimental value, respectively, both in gas phase and in dissolved phase at the B3LYP/6-311++G** level. At the RHF/6-311++G** level, the C5-N6 and C1-N6 bond lengths are 0.029 Å and 0.041 Å smaller than the C-N experimental value, respectively, both in the gas phase, water and ethanol. From
Table 1a, we observed that C-N bond lengths on the Pyrimidine ring vary slightly. This variation of
Fig.1: Optimized Structure of Pyrimethamine [32,33].
the C-N bond lengths suggests that there is an extended pi-electron delocalization over the pyrimidine moiety.
The bond angles vary from 0.1 to 0.2 degree(s)
in both RHF and DFT levels of theories with the 6-
The African Review of Physics (2013) 8:0062 466
311++G** basis set (Table 1b). The six member
angles vary slightly as we move from gas phase to
carbon ring (Phenyl) and the other ring with two of
solution phase. Most of the bond angles increases
the carbon atoms replaced by Nitrogen atoms
as we move from gas medium to water and ethanol
(pyrimidine) possibly give added stability to the
with the increase more significant in ethanol. The
molecule. The nitrogen atoms (N11, N12, N14 and
bond angles (Table 1b) at the RHF/6-311++G**
N17) play a major role in the electron density
and at the B3LYP/6-311++G** are approximately
configuration. Once dissolved (in water and
equal to the experimental results [34,35,36]. The
ethanol) the bond angles remain unaffected at
RHF/6-311++G** results are in better accord with
RHF/6-311++G** level of theory while at
the experimental results.
B3LYP/6-311++G** level of theory, the bond
Table 1a: Optimized geometrical parameters – Bond lengths of Pyrimethamine molecule in different mediums obtained by RHF and B3LYP methods employing 6- 311++G** basis sets and their corresponding experimental values in gas.
B3LYP/6-311++G**
1.3244 1.3395 1.3394 1.3394
1.3185 1.3372 1.3372 1.3371
R(C1- N19) 1.3582
1.3582 1.3691 1.3693 1.3693
1.3187 1.3344 1.3344 1.3344
R(C3-N17) 1.3547
1.3547 1.3678 1.3679 1.3680
1.3305 1.3429 1.3429 1.3429
1.4131 1.4199 1.4198 1.4199
1.3839 1.3974 1.3974 1.3974
1.4967 1.4922 1.4921 1.4921
R(C5-C24) 1.5101
1.5101 1.5113 1.5112 1.5111
1.3911 1.4019 1.4019 1.4019 1.402
1.3914 1.4020 1.4020 1.4020 1.402
R(C8-C10) 1.3853
1.3853 1.3933 1.3932 1.3932
R(C9-C12) 1.3854
1.3854 1.3933 1.3933 1.3933
R(C12-C14) 1.3812
1.3812 1.3909 1.3909 1.3909
R(C24-C25) 1.5329
1.5329 1.5383 1.5382 1.5382
R(C10-C14) 1.3813
1.3813 1.3910 1.3911 1.3911
R(C14-Cl22) 1.7450
1.7450 1.7594 1.7593 1.7593
R(C8-H11) 1.0752
1.0752 1.0843 1.0843 1.0843 1.082
R(C9-H13) 1.0756
1.0756 1.0844 1.0844 1.0844
R(C10-H15) 1.0737
1.0737 1.0827 1.0827 1.0827
R(C12-H16) 1.0737
1.0737 1.0827 1.0827 1.0827
R(C24-H26) 1.0849
1.0849 1.0939 1.0940 1.0940
R(C24-H27) 1.0821
1.0821 1.0910 1.0910 1.0910
R(C25-H28) 1.0858
1.0858 1.0932 1.0932 1.0932
R(C25-H29) 1.0836
1.0836 1.0917 1.0917 1.0917
R(C25-H30) 1.0863
1.0863 1.0937 1.0937 1.0937
R(N17-H18) 0.9929
0.9929 1.0076 1.0077 1.0077
R(N17-H23) 0.9914
0.9914 1.0064 1.0065 1.0065
The African Review of Physics (2013) 8:0062 467
R(N19-H20) 0.9924 0.9924
0.9924 1.0065 1.0066 1.0066
R(N19-H21) 0.9924
0.9924 1.0066 1.0067 1.0067
Bond Lengths (R) are given in Armstrong (Å) and atoms labeling according to Fig. 1.
Table 1b: Optimized geometrical parameters – Bond angles of Pyrimethamine molecule in different mediums obtained at RHF and B3LYP methods by employing 6- 311++G** basis sets and their corresponding experimental values in gas.
B3LYP/6-311++G**
126.5661 126.5661 126.5813
126.5887 126.5882
111.4042 116.4042 116.5302
116.5277 116.5253
117.0147 117.0147 116.8667
116.8614 116.8640
116.0653 116.0653 116.1734
116.1680 116.1686
116.5644 116.5644 116.3674
116.3618 116.3625
116.8374 116.8374 116.7324
116.7293 116.7302
122.4486 122.4486 122.4422
122.4458 122.4461
121.4737 121.4737 121.3714
121.3729 121.3721
115.0879 115.0879 115.5793
115.5766 115.5759
120.7408 120.7408 120.4696
120.4700 120.4766
124.1710 124.171 123.9511
123.9534 123.9475
122.4880 122.488 122.2910
122.2916 122.2910
122.6870 122.6870 122.6697
122.6674 122.6722
121.0529 121.0529 120.9668
120.9751 120.9811
120.9544 120.9544 121.0545
121.0507 121.0451
117.9829 117.9829 117.9685
117.9640 117.9639
121.3749 121.3749 121.3827
121.3856 121.385
121.4024 121.4024 121.4082
121.4102 121.4106
119.1411 119.1411 119.0947
119.0966 119.0967 119.80
119.1812 119.1812 119.1240
119.1256 119.1266
120.9153 120.9153 121.0186
121.0148 121.0139
111.9558 111.9558 112.0507
112.0684 112.0646
119.5466 119.5466 119.4919
119.492 119.4934
119.5380 119.5380 119.4894
119.4924 119.4926
119.4315 119.4315 119.344
119.3386 119.3393
119.1936 119.1936 119.273
119.2755 119.2754
119.4153 119.4153 119.3155
119.3184 119.3161
119.1812 119.1812 119.2758
119.2709 119.2728
120.6316 120.6316 120.7266
120.7265 120.7255
120.1872 120.1872 120.1492
120.1476 120.1476
120.6491 120.6491 120.7408
120.7385 120.7389
120.2098 120.2098 120.1644
120.1648 120.1644
110.5948 110.5948 110.4605
110.4617 110.4679
109.3066 109.3066 109.1122
109.1033 109.1035
The African Review of Physics (2013) 8:0062 468
109.8061 109.8061 109.8103
109.8094 109.8121
107.8799 107.8799 107.7345
107.7305 107.7264
110.1473 110.1473 110.5216
110.5131 110.5133
110.7270 110.7270 110.4946
110.5022 110.5002
111.1655 111.1655 111.1290
111.1442 111.1429
116.7362 116.7362 116.5334
116.4973 116.4914 113.90
120.0663 120.0663
119.5782 119.5473 119.5409
117.1351 117.1351
117.3233 117.3359 117.3108
116.8222 116.8222
116.8992 116.9110 116.8888
118.0941 118.0941
118.3329 118.3405 118.3083
118.1127 118.1127
117.9775 117.9435 117.9371
108.4609 108.4609
108.4215 108.4248 108.4265 109.01
107.9126 107.9126
107.9423 107.9361 107.9357
108.3325 108.3325
108.2345 108.2224 108.2243
Bond Angles (A) are in degrees (˚) and atoms labeling according to Fig.1.
3.2. Optimized geometric properties of
experimental value at the B3LYP/6-311++G**
sulfadoxine
level and at the RHF/6-311++G** level, it is 0.114 Å smaller both in the gas phase and dissolved in
The geometric parameters of Sulfadoxine molecule
water and ethanol. From Table 2a, we also
in gas phase, water and ethanol are listed in Table
observed that C-N bond lengths on the Pyrimidine
2a. The calculated bond lengths at RHF/6-
ring vary slightly. This variation of the C-N bond
311++G** level are slightly smaller (ranging from
lengths also suggests that there is an extended pi-
0.01Å to 0.04Å) than their corresponding values
electron delocalization over the pyrimidine moiety.
obtained at the B3LYP/6-311++G** level. In
The bond angles vary from 0.1 to 0.2 degrees in
solution (water and ethanol) the bond lengths
both RHF and DFT levels of theories with the 6-
remain unaffected at the RHF/6-311++G** level of
311++G** basis set (Table 2b). The Phenyl group
theory and at the B3LYP/6-311++G** level of
and the pyrimidine possibly give added stability to
theory. The RHF/6-311++G** and B3LYP/6-
the molecule. The Nitrogen atoms (N4, N17, N18
311++G** bond lengths are approximately equal to
and N32) and the oxygen atoms (O6, O7, O20 and
the experimental values [34,35,36]. The B3LYP/6-
O21) play a major role in the electron density
311++G** theoretical calculated values are in
configuration. In the water and ethanol dissolved
better agreement with experimental values than
state, the bond angles remain unaffected both at the
their corresponding RHF/6-311++G** theoretical
RHF/6-311++G** level and at the B3LYP/6-
calculated values. For example in the Pyrimidine
311++G** level of theory as we move from gas
ring, the C1-N2 bond length is 0.105 Å, which is
phase to water and to ethanol. The bond angles
smaller than the C-N experimental value at the
(Table 2b) at the RHF/6-311++G** and at the
B3LYP/6-311++G** level and at the RHF/6-
B3LYP/6-311++G** are approximately equal to
311++G** level. It is 0.124 Å smaller than the C-N
the experimental results [34,35,36].
experimental value respectively both in the gas
phase and solution in water and ethanol. The C1-N6 bond length is 0.097 Å smaller than the
The African Review of Physics (2013) 8:0062 469
Table 2a: Optimized geometrical parameters – Bond lengths of Sulfadoxine molecule in different medium obtained at RHF and B3LYP methods by employing 6- 311++G** basis sets and their corresponding experimental values.
Bond Lengths (R) are given in Armstrong (Å) and atoms labeling according to Fig. 2.
The African Review of Physics (2013) 8:0062 470
Table 2b: Optimized geometrical parameters – Bond angles of Sulfadoxine molecule in different medium obtained at RHF and B3LYP methods by employing 6- 311++G** basis sets and their corresponding experimental values.
119.9897 119.9897
119.9464 119.9464
119.9764 119.9764
119.6635 119.6635
120.4593 120.4593
119.8619 119.8619
119.8433 119.8433
119.7726 119.7726
119.7614 119.7614
115.6438 115.6438
120.2143 120.2143
120.0275 120.0275
119.8771 119.8771
120.2932 120.2932
120.4659 120.4659
119.1203 119.1203
A(C30-N33-H34) 116.2733
116.2733 116.2733
A(C30-N33-H35) 116.3096
116.3096 116.3096
A(H34-N33-H35) 113.0062
113.0062 113.0062
A(H11-C10-H12) 110.4143
110.4143 110.4143
A(H11-C10-H13) 110.3132
110.3132 110.3132
A(H12-C10-H13) 109.3598
109.3598 109.3598
127.2026 127.2026
A(N2-C3-N18) 118.8657
118.8657 118.8657
A(N2-C1-H7) 116.5600
116.5599 116.5599
A(N6-C1-H7) 116.2354
116.2354 116.2354
116.1156 116.1156
116.5139 116.5139
122.4483 122.4483
118.6516 118.6516
122.0482 122.0482
120.3781 120.3781
120.4724 120.4724
120.3587 120.3587
123.8947 123.8947
117.8637 117.8637
A(N6-C5-O8) 120.0855
120.0855 120.0855
119.2644 119.2644
The African Review of Physics (2013) 8:0062 471
116.3076 116.3076
A(O8-C10-H11) 105.2619
105.2619 105.2619
A(O8-C10-H12) 110.7611
110.7611 110.7611
A(O8-C10-H13) 110.6802
110.6802 110.6802
A(O9-C14-H15) 106.4717
106.4717 106.4717
A(O9-C14-H16) 110.5699
110.5699 110.5699
A(O9-C14-H17) 110.7178
110.7178 110.7178
A(H15-C14-H16) 109.5985
109.5985 109.5985
A(H15-C14-H17) 109.5906
109.5906 109.5906
A(H16-C14-H17) 109.8313
109.8313 109.8313
A(C3-N18-H19) 116.9503
116.9503 116.9503
A(C3-N18-S20) 127.4524
127.4525 127.4525
A(H19-N18-S20) 112.8406
112.8406 112.8406
A(N18-S20-O21) 109.3527
109.3527 109.3527
A(N18-S20-O22) 101.7625
101.7625 101.7625
A(N18-S20-C23) 106.7853
106.7853 106.7853
A(O21-S20-O22) 120.5877
120.5877 120.5877
A(O21-S20-C23) 108.7410
108.7410 108.7410
A(O22-S20-C23) 108.7118
108.7118 108.7118
A(S20-C23-C24) 119.5720
119.5720 119.5720
A(S20-C23-C25) 120.2006
120.2006 120.2006
Bond Angles (A) are in degrees (˚) and atoms labeling according to Fig. 2.
4. Atomic Charges, Polarizability and Hyper-
Hence, it is appropriate to consider the charges
calculated by CHELPG scheme of Breneman instead of Mulliken population analysis. Within a
Atomic net charges are not quantum mechanical
molecular system, atoms can be treated as a
(QM) observables, and they cannot be determined
quantum mechanical system. On the basis of the
directly with QM calculations or by experiments.
topology of the electron density, the atomic charges
Different methods exist for the estimation of
in the molecule can be explained.
atomic charges of the molecular system. Basically,
Polarizability is a property that depends on the
the atomic charges are best derived by a least
second derivative of the energy with respect to the
squares fit to the electrostatic potential (ESP),
applied electric field. It gives information about the
calculated in a large number of points around the
molecule of interest [38].
Polarizability plays an important role in the
The electrostatic potential derived charges
understanding of a large variety of physical
using the CHELPG scheme of Breneman [39] at
different atomic positions of Pyrimethamine and
experimental difficulties obtaining reliable results
Sulfadoxine molecules at RHF/6-311++G** and
of polarizabilities justify the need of accurate
B3LYP/6-311++G** levels of theories have been
theoretical results. In some cases, these results are
calculated. The Mulliken population analysis
partitions the charges among the atoms of a
polarizabilities.
molecule by dividing orbital overlap evenly
components, the average polarizability and the
between two atoms. Whereas, the electrostatic
potential derived charges assign point charges to fit
Pyrimethamine and Sulfadoxine obtained at
the computed electrostatic potential at a number of
RHF/6-311++G** level and B3LYP/6-311++G**
points on or near the Van der Waal surface. This
level of theories are listed in Tables 3 and 4,
sort of analysis is commonly used to create input
respectively. All the six polarizability tensor
charges for molecular mechanics calculation.
The African Review of Physics (2013) 8:0062 472
components of Pyrimethamine and Sulfadoxine
the anisotropy for the molecule at all levels of
molecules αxx, αxy, αyy, αxz αyz and αzz components
theory. We can also see that the inclusion of
changes significantly at the RHF/6-311++G** level
B3LYP/6-311++G**
polarizability < α> and the anisotropy γ in gas
considered here (although they do not follow any
phase and in solution phase. We equally observe
regular pattern).
that the effect of inclusion of electron correlation
In the case of Pyrimethamine, the polarizability
increases < α > by 17.30 percent for the gas phase,
tensor components do not show any change in
17.17 percent when dissolved in water and 17.31
going from gas phase to solution phase at the
percent in ethanol whereas γ increases by 45.50
RHF/6-311++G** level but polarizability tensor
percent for the gas phase, 45.56 percent when
components change slightly at the B3LYP/6-
dissolved in water and 45.49 percent when in
311++G** level. From Table 3, we can see that
tensor αxx is responsible for the greatest
contribution both in the average polarizability and Table 3: Polarizability tensors, average polarizability and anisotropy (x 10-12 esu) of Pyrimethamine using RHF and B3LYP methods by employing 6-311++G** basis set.
B3LYP/6-311++G**
polarizability < α> and the anisotropy γ in gas
polarizability tensor components of Sulfadoxine do
phase and in solution phase. We equally observed
not show any change in going from gas phase to
that the effect of inclusion of electron correlation
solution phase at the RHF/6-311++G** level but
increases < α> by 4.42 percent for the gas phase,
polarizability tensor components change slightly at
0.85 percent when dissolved in water or in ethanol
the B3LYP/6-311++G** level. From Table 4, we
whereas γ increases by 38.80 percent for the gas
can see that the tensor αxx is responsible for the
phase, 26.01 percent when dissolved in water or in
polarizability and the anisotropy for the molecule at
all levels of theory. We can also see that the inclusion of electron correlation affects the average Table 4: Polarizability tensors, average polarizability and anisotropy (x 10-12 esu) of Sulfadoxine using RHF and B3LYP methods by employing 6-311++G** basis set.
B3LYP/6-311++G**
The African Review of Physics (2013) 8:0062 473
The polarizability and hyperpolarizability of a
and when dissolved in ethanol, whereas βzzz
molecule are presented as the output in the standard
component gives the highest contribution when
orientation in triangular and tetrahedral order,
dissolved in water. The first molecular hyper-
respectively. Hyper-polarizability is very sensitive
polarizability is greater at the B3LYP/6-311++G**
to molecular structure and highly dependent on the
compared to the RHF/6-311++G**, both in gas
choice of the basis set [43,44]. Tables 5 and 6 show
phase and in solution phase.
the diagonal hyper-polarizabilities and the first
In the case of Sulfadoxine, the component βzzz
molecular hyper-polarizability of Pyrimethamine
of the hyper-polarizability gives the highest
and Sulfadoxine molecules. Since the values of the
contribution to molecular hyper-polarizability at
first molecular hyper-polarizability are given in
the RHF/6-311++G** level in both gas phase and
atomic units, we have thus converted the values in
solution phase. At the B3LYP/6-311++G** level,
electrostatic units.
the component βyyy of the hyper-polarizability gives
In the case of Pyrimethamine, the component
the highest contribution of the first molecular
βxxx of hyper-polarizability gives the highest
hyper-polarizability when dissolved in water and
contribution to the
ethanol and βzzz component gives the highest
polarizability at the RHF/6-311++G** level in both
contribution in gas phase. The first molecular
gas phase and solution phase. At the B3LYP/6-
hyper-polarizability is also greater at the B3LYP/6-
311++G** level, the component βxxx of the hyper-
311++G** compare to the RHF/6-311++G** both
polarizability gives the highest contribution of the
in gas phase and in solution phase.
first molecular hyper-polarizability in gas phase
Table 5: Components of diagonal hyper-polarizabilities and first molecular hyper-polarizabilities (x 10-33esu) of Pyrimethamine using RHF and B3LYP methods by employing 6-311++G** basis set.
B3LYP/6-311++G**
-10234.486 698.980
-510.151 11323.963
Table 6: Components of diagonal hyper-polarizabilities and molecular first hyper-polarizability (x 10-33esu) of Sulfadoxine using RHF and B3LYP methods by employing 6-311++G** basis set.
B3LYP/6-311++G**
-18845.976 -18845.976 -25100.063 2475.790
-11599.634 -11599.634 -15575.985 -18058.020 -18058.020
-28028.844 -28028.844 -37368.636 -2209.536
The African Review of Physics (2013) 8:0062 474
5. Conclusion
F. Kajzar, K. S. Lee and A. K. Jen, Adv.
Poly. Sc. 161, 1 (2003).
We observed that some charge transfer takes place
V. Krishnakumar and R. Nagalakshmi,
in going from gas phase to solution medium at the
Physica B, 403, 1863 (2008).
B3LYP/6-311++G** level of theory. This charge
H. Alyar, Rev. Adv. Mater. Sci. 34, 79
transfer is more significant in the case of water than
in the case of ethanol. The polarizability of these
Y. Zhang and R. H. Holm, J. Amer. Chem.
molecules changed slightly following their solution
Soc. 125, 3910 (2003).
in different media, in comparison to gas phase, at
D. S. Chemla and J. Zyss, Nonlinear Optical
the B3LYP/6-311++G** level of theory.
Properties of Organic Molecules and
The high polarizability values of the molecules
Crystals (Academic Press, Orlando, 1987).
revealed that the electrostatic and dispersion
P. N. Prasad and D. J. Williams, Intro-
contribution influence considerably the interaction
duction to Nonlinear Optical Effects in
of these molecules with other molecules. We also
Molecules and Polymers (Wiley, New York
concluded that an appropriate treatment of the
electron correlation is of fundamental importance
J. Zyss, Molecular Nonlinear Optics
in order to obtain accurate estimates for the
(Academic Press, Boston, USA, 1994).
electronic contributions to polarizabilities and
N. Venkatram, M. A. Akundi and D.
hyper-polarizabilities.
Narayana Rao, Optical Express 13, 867
The variation of the C-N bond lengths suggests
an extended pi-electron delocalization over the
G. W. Ejuh and L. S. Taura, J. Nigerian
pyrimidine moiety, which is responsible for the non
Association of Mathematical Phys. 20(1),
linearity of these molecules.
Due to the large first hyper-polarizability of
P. Gunter, Nonlinear Optical Effects and
these molecules, we concluded that these molecules
Materials (Springer-Verlag, Berlin, 2000).
have the potential to produce second order optical
D. R. Kanis, M. A. Ratner and T. J. Marks,
effects much larger than those obtainable with
Chem. Rev. 94, 195 (1994).
inorganic materials which suggests potential for
P. Poornesh, G. Umesh, P. K. Hegde, M. G.
applications of these molecules in the development
Manjunatha, K. B. Manjunatha and A. V.
of non linear materials. Hence these molecules,
Adhikari, Applied Phys. B. 97, 117 (2009).
which are Malaria drugs also, have application in
L. T. Cheng, W. Tam, S. H. Stevenson, G.
electro optics devices, photorefractive materials,
R. Meredith, G. Rikken and S. R. Marder, J.
second harmonics generation, etc.
Phys. Chem. 95, 1063 (1991).
Due to the lack of existing experimental or
P. S. Liyanage, R. M. De Silva and K. M. N.
theoretical results of the hyper-polarizability of
De Silva, J. Mol. Struct. (Theochem), 639,
these molecules, we are optimistic that our results
will provide a benchmark study for these
K. S. Thanthiriwatte and K. M. N. De Silva,
molecules, and serve as a reference for other
J. Mol. Struct. 617, 169 (2002).
A. D. Tillekaratne and K. M. N. De Silva, J.
Mol. Struct. 638, 169 (2003).
Y. Atalay, D. Avci, and A. Basoglu, Struct.
We want to thank the CSIR (Council of Scientific
Chem. 19, 239 (2008).
and Industrial Research), New Delhi, for providing
C. R. Moylam, R. J. Twieg, V. Y. Lee, S. A.
funds with which the Gaussian 03W software was
Swanson, K. M. Betteron and R. D. Miller,
purchased through Prof. A. N. Singh, Banaras
J. Chem. Soc. 115, 12599 (1993).
Hindu University, Varanasi, India.
V. P. Rao, Y. M. Cai and A. K. Y. Jen, J.
We equally want to thank Prof. A. N. Singh, for
Chem. Soc. Chem. Commun. 14, 1689
providing us with the Software and initiating our
research in this area.
S. Zafar, Z. H. Khan and M. S. Khan,
Canadian J. Pure and Applied Sc. 6(1), 1827
References
H. Alyar, M. Bahat, E. Kasap and Z.
Y. Shi, C. Zhang, J. H. Bechtel, L. R.
Kantarci, Czech. J. Phys. 56, 349 (2006).
Dalton, B. H. Robinson and W. H. Steier,
Science 288, 119 (2000).
The African Review of Physics (2013) 8:0062 475
H. Soscun, O. Castellano, Y. Berudez, C.
K. H. Hellwege and A. M. Hellwege,
Landolt-Bornstein, Atom. and Mol. Phys.
Alvarado, J. Mol. Struct. 592, 19 (2002).
(Springer-Verlag, Berlin, 1976).
H. Li, K. Han, X. Shen, Z. Lu, Z. Huang, W.
G. Roussy, J. Mol. Spec. 118, 180 (1986).
Zhang, Z. Zhang and L. Bai, J. Mol. Struct.
B. A. Saleh, H. A. Abood, R. Miyamoto and
(Theochem), 767, 113 (2006).
M. Bortouzzi, J. Iranian Chem. Soc. 8(3),
B. Champagne and M. Spassova, Chem.
Phy. Lett. 471, 111 (2009).
B. Mannfors, K. Palmo and S. Krimm, J.
G. W. Ejuh, J. M. Ndjaka and A. N. Singh,
Mol. Struct. 556, 1 (2000).
Canadian J. Pure and Applied Sc. 5(2), 1591
C. M. Breneman and K. B. Wiberg, J.
Comput. Chem. 11, 361 (1990).
A. Andrea, Adv. Phys. Chem. (Hindawi
A. D. Buckingham, Adv. Chem. Phys. 12,
Publishing Cooperation, 2013).
M. J. Frisch, G. W. Trucks, H. B. Schlegel,
E. F. Archhibong and A. J. Thakkar, J. Phys.
G. E. Scuseria, M. A. Robb, J. R.
Rev. A 44, 5478 (1991).
Cheeseman, J. A. Montgomery, Jr. T.
M. A. Castro and S. J. Canuta, J. Phys. Rev.
Vreven, K. N. Kudin, J. C. Burant, J. M.
B 26, 4301 (1993).
Millam, S. S. Iyengar, J. Tomasi, V. Barone,
D. V. Fernando, A. S. David, T. Yoshinari,
B. Mennucci, M. Cossi, G. Scalmani, N.
A. Xavier, R. Angel, G. L. Steven and J. R.
Rega, G. A. Petersson, H. Nakatsuji, M.
John, J. Chem. Phys. 133, 034111 (2010).
Hada, M. Ehara, K. Toyota, R. Fukuda, J.
J. Kobus, D. Moncrieff and S. Wilson, J.
Hasegawa, M. Ishida, T. Nakajima, Y.
Phys. B. Atom. Mol. and Optical Phys.
Honda, O. Kitao, H. Nakai, M. Klene, X. Li,
37(3), 571 (2004).
J. E. Knox, H. P. Hratchian, J. B. Cross, C.
Adamo, J. Jaramillo, R. Gomperts, R. E.
Stratmann, O. Yazyev, A. J. Austin, R.
Cammi, C. Pomelli, J. W. Ochterski, P. Y.
Ayala, K. Morokuma, G. A. Voth, P.
Received: 5 October, 2013
Zakrzewski, S. Dapprich, A. D. Danniels,
Accepted: 17 January, 2014
M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian Inc., Wallingford CT. (2004).
A. D. Becke, Phys. Rev. A. 38, 3098 (1988).
C. Lee, W. Yang and R. G. Parr, Phys. Rev.
B 37, 785 (1988).
S. H. Vosko, L. Wilk and Nusair, Canadian
J. Phys. 58, 1200 (1980).
G. W. Ejuh, J. M. Ndjaka and A. N. Singh,
Global J. Pure & Applied Sc. and Tech.
1(2), 30 (2011).
G. W. Ejuh, J. M. Ndjaka and A. N. Singh,
African Rev. Phys. 6(3), 21 (2011).
G. Herzberg, Electronic Spectra and Electronic
Structure
Polyatomic
Molecules (Van Nostrand, New York, 1966).
Source: http://www.aphysrev.org/index.php/aphysrev/article/viewFile/832/340
Vivere e lavorare in Svizzera Il Paese e i suoi abitanti Entrata e dimora Vivere in Svizzera Lavorare in Svizzera Sicurezza sociale Ufficio federale della migrazione UFM Prefazione Le informazioni sono destinate alle persone desiderose di stabilirsi e di lavorare in Svizze-ra. Ringraziamo gli uffici e i servizi che hanno contribuito alla presente pubblicazione. Non esitate a comunicarci modifiche e aggiornamenti. Per ulteriori informazioni, per ordinare altri dossier o per fissare un colloquio personale con uno dei nostri consulenti, ci si può rivolgere a:
Medical data is for informational purposes only. You should always consult your family doctor or one of our referral physicians. Gouty Arthritis by Anthony di Fabio The Roger Wyburn-Mason and Jack M. Blount Foundation for the Eradication of Rheumatoid Disease AKA The Arthritis Trust of America®, 7376 Walker Road, Fairview, Tn 37062

