Pyrazole pendant tetraazamacrocyclic ligands for possible medicinal applications
State University of New York College at Buffalo - Buffalo State College
Pyrazole Pendant Tetraazamacrocyclic Ligands for
Possible Medicinal ApplicationsAnjuli Bhandari
Buffalo State College,
[email protected]
Advisor
M. Scott Goodman, Ph.D., Chair and Professor of Chemistry
First Reader
M. Scott Goodman, Ph.D., Chair and Professor of Chemistry
Second Reader
William Durfee, Ph.D. Professor of Chemistry
Third Reader
Jinseok Heo, Ph.D. Associate Professor of Chemistry
Department Chair
M. Scott Goodman, Ph.D., Professor of Chemistry
To learn more about the Chemistry Department and its educational programs, research, andresources, go to .
Recommended CitationBhandari, Anjuli, "Pyrazole Pendant Tetraazamacrocyclic Ligands for Possible Medicinal Applications" (2016).
Forensic Science Theses.
Paper 11.
Follow this and additional works at:
Part of the , and the
Pyrazole Pendant Tetraazamacrocyclic
Ligands for Possible Medicinal Applications
An Abstract of a Thesis
Submitted in Partial Fulfillment
of the Requirements
for the Degree of
Master of Science
State University of New York
College at Buffalo
Department of Chemistry
ABSTRACT OF THESIS
Pyrazole Pendant Tetraazamacrocyclic Ligands for Possible Medicinal Applications
Gadolinium is the element typically used for MRI contrast agents, which are sometimes
injected prior to performing an MRI. The element accumulates in inflamed areas, which
indicates where there may be an infection or a tumor. However, gadolinium is more toxic to the
body than preferred and the potential long-term effects have been studied in individuals who
experience health issues.
Complexes containing metal ions that are naturally occurring have been proposed to replace
gadolinium use. Complexing a metal ion (i.e. copper(II) or iron(II)) to a macrocyclic ligand that
has a high water exchange rate would potentially be able to replicate gadolinium–based dye
conditions while also having a higher excretion rate and lower toxicity. A research project was
proposed based on the findings of Morrow and co-workers
that an iron-containing complex
could function as a PARACEST MRI contrast agent.
Tetraazamacrocyclic ligands such as cyclam and cyclen are useful macrocyclic ligands for
new research in drug therapy and imaging techniques. The addition of pyrazole pendant arms
has been proposed as a possible option to improve their ligating properties. Appending these
groups to the tetraazamacrocyclic ligands cyclam and cyclen was the initial focus of the research.
These ligands were subsequently used to attempt to form stable complexes using a variety of
metal ions and solvents. All new compounds were characterized using a combination of NMR,
mass spectrometry, and X-ray crystallography. Compounds of the type synthesized show
promise in one day being a strong contender for use in MRI contrast and possibly therapy for
cancerous tumors.
State University of New York
College at Buffalo
Department of Chemistry
Pyrazole Pendant Tetraazamacrocyclic
Ligands for Possible Medicinal Applications
Submitted in Partial Fulfillment
of the Requirements
for the Degree of
Master of Science
M. Scott Goodman, PhD
Chemistry Department Chair and Professor
Chairperson of the Committee
Kevin J. Railey, Ph.D.
Associate Provost and Dean of the Graduate School
THESIS COMMITTEE
M. Scott Goodman, Ph.D.
Chemistry Department Chair, Professor of Chemistry
Chairperson of the Committee/Thesis Advisor
William S. Durfee, Ph.D.
Professor of Chemistry
Jinseok Heo, Ph.D.
Associate Professor of Chemistry
Thank you my research advisor, Dr. Scott Goodman for the wealth of knowledge you
have provided me. With your guidance I have become familiar with organic chemistry and
would not be heading in the direction I am without this experience.
I would like to thank my committee members, Dr. William Durfee and Dr. Jinseok Heo
for your time and suggestions.
Also, thank you to Dr. Alex Nazarenko for providing X-ray crystallography that was used
throughout my thesis.
I would also like to thank, Danielle Precourt, Elisabeth Barone and Kayleigh Bemisderfer
for your continued support over these last two years. Lastly, thank you to my family and
Nicholas, who has continuously encouraged me, pushed me, and motivated me to be the best I
can be and to always try harder. Thank you for all of your continued support over these past six
Table of Contents
Abstract Title Page . i
Abstract of Thesis . ii
Title Page . iii
Committee Signatory . iv
Acknowledgements .v
Chapter 1: Introduction .1
1.0 Introduction-Tetraazamacrocycles .1
1.03 Cyclen Macrocycle in Medicinal Applications .4
1.04 Importance of Macrocyclic Metal Ion Complexes in Medicinal Applications .5
1.05 Optimum Binding of Metal Ions .6
1.06 Topology .6
1.07 Rigidity .8
1.08 Pyrazoles .9
1.09 Pendant Arms .10
1.10 Coordination Number .10
1.11 Magnetic Resonance Imaging .11
1.12 MRI Contrast Agents .12
1.13 Gadolinium Paramagnetic Relaxation .15
1.14 ParaCEST .16
1.15 Magnetization Transfer Effect .16
1.16 Previously Synthesized ParaCEST Complexes .17
1.17 Macrocyclic Ligands use as MRI Contrast Agent .18
Chapter 2: Materials and Methods .21
2.1 Materials .21
Chapter 3: Results and Discussion .28
3.1 Pyrazoles as Pendant Arms .28
3.2 Tetraethylpyrazolyl Cyclam (
PzCm) .29
3.3 Tetramethylpyrazolyl Cyclen (
PzCn) .37
3.4 Tetradimethylpyrazolyl Cyclam (
DiMePzCm) .45
3.5 Tetradimethylpyrazolyl Cyclen (
DiMePzCn) .54
3.6 Comparison of Ligands .65
3.7 PĪ Space Group .70
3.8 Synthetic Considerations .70
3.9 Metal Ion Complex Attempts .71
Chapter 4: Conclusions and Future Directions.73
4.0 Conclusions .73
List of Figures
1. The structure of 1,4,8,11-tetraazacyclotetradecane (cyclam) . 3
2. The structure of 1,4,7,10-tetraazacyclododecane (cyclen) . 4
3. Binding of donor atoms in chelate versus monodentate ligands . 8
4. The chemical structure of pyrazole . 9
5. The chemical structure of 3,5-dimethylpyrazole . 10
6. Square prismatic, left, versus antiprismatic, right, example . 11
7. Generated MRI image . 12
8. MRI with no contrast, left, versus, right . 13
9. 1,4,7-tris(carbamoylmethyl)-1,4,7-triazzacyclcononane ParaCEST
10. Rigidified ligand of 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid
(DOTA) adapted from Chong
et al . 19
11. Chemical structure of 1-(2-bromoethyl)pyrazole (a) and 1-(2-bromoethyl)-3,5-
dimethylpyrazole (b) . 28
12. Chemical structure of 1-(2-chloroethyl)pyrazole (a) and 1-(2-chloroethyl)-
3,5-dimethylpyrazole (b) . 28
13. The structure of
PzCm . 29
14. Mass spectrum of
PzCm . 31
15. 13C NMR spectra of
PzCm . 32
16. Structure of
PzCm for NMR assignment . 32
17. The 1H NMR spectra of
PzCm . 33
18. IR spectra of
PzCm . 34
19. X-ray crystallography of
PzCm. Hydrogen atoms are omitted for clarity. 35
20. The structure of
PzCn . 38
21. The 1H NMR spectra of
PzCn . 39
22. The structure of
PzCN labeled for NMR assignment . 40
23. 13C NMR spectra of
PzCn . 40
24. Mass spectrum of
PzCn . 41
25. IR spectrum of
PzCn. 42
26. X-ray crystallography of
PzCn. Hydrogen atoms are omitted for clarity. 43
27. The structure of
DiMePzCm . 46
28. The 1H NMR spectrum of
DiMePzCm . 47
29. Labeled structure of
DiMePzCm . 48
30. 13C NMR spectrum of
DiMePzCm . 48
31. High resolution mass spectrum of
DiMePzCm actual (top) versus
theoretical (bottom) . 49
32. IR spectrum of
DiMePzCm . 50
33. X-ray crystallography of
DiMePzCm. Hydrogen atoms are not shown. 51
34. Structure of
DiMePzCn . 55
35. The 1H NMR spectrum of
DiMePzCn . 56
36. The labeled structure of
DiMePzCn for NMR assignment . 57
37. 13C NMR spectrum of
DiMePzCn . 57
38. Fragmented mass spectrum of
DiMePzCn . 59
39. Molecular ion mass spectrum of
DimePzCn . 60
40. High resolution mass spectrum of
DiMePzCn doubly charged . 61
41. IR spectrum of
DiMePzCn . 62
42. X-ray crystallography of
DiMePzCn. Hydrogens are omitted for clarity. 63
43. Crystal structures of
PzCm and
DiMePzCm for comparison . 67
44. Crystal structures of
PzCn and
DiMePzCn for comparison . 69
List of Tables
1.
PzCm crystallographic data . 36
2.
PzCm bond lengths . 36
3. Selected bond angles for
PzCm. 37
4.
PzCn crystallographic information . 44
5. Bond lengths of
PzCn . 44
6. Selected bond angles for
PzCn . 45
7.
DiMePzCm crystallographic information . 52
8. Bond lengths of
DiMePzCm . 53
9. Selected bond angles of
DiMePzCm . 54
10.
DiMePzCn crystallographic information . 63
11. Bond lengths of
DiMePzCn . 64
12. Selected bond angles for
DiMePzCn . 65
13. Characterization data for
PzCm, PzCn, DiMePzCm, and
DiMePzCn . 66
14. Attempted metal ion reactions. 72
Chapter 1. Introduction
1.0 Introduction–Azamacrocycles
Macrocyclic ligands are polydentate ligands. This refers to the fact that more than one
atom is capable of forming bonds to metal ions is present. Macrocyclic polydentate
ligands typically possess at least 3 donor atoms that will assist in the binding of a metal
ion. Generally, macrocycles contain 8 or more total atoms in the ring, but this number can
vary. A compound with only 8 atoms will have a more difficult time complexing and
retaining a larger ion than a compound with 12 or 14 atoms, but might be more suitable
for a smaller metal ion. Once the metal is bound to the ligand, it will be held in place in
the center of the complex by the pre-organized structure of the ligand.1,2 The nature of
this complex depends on the number of donor atoms as well as size of the macrocycle
itself. The macrocyclic backbone can also be either saturated or unsaturated. If the
complex is unsaturated the compound will be less flexible where a saturated compound
will be more flexible. Lack of flexibility is an advantage here because it will retain the
metal ion in the "cavity" of the ligand and there will be less chance of dissociation
occurring of the metal-ligand complex.1
Macrocyclic ligands have been of research interest since the 1960's but interest has
increased exponentially due to the stability and variability of these ligands. Busch, Curtis,
and Pedersen initiated research of macrocyclic ligands during the 1970's, and around
1980 the focus shifted more towards research on biomedical applications of these
macrocyclic compounds.3 Macrocycles can be found in naturally occurring bodily
processes (e.g. porphyrin ring of heme and photosynthesis) and can also be found in some
synthetic dyes.1,2 It has become evident throughout the course of research and continuing
research that macrocycles have value in multiple areas of medicine.
Polyazamacrocycles are of particular interest when studying metal ion complexes due
to the variability of applications and increased stability when compared with linear
chelates. When pendant groups are ligated to the azamacrocycles, a metal ion can be
coordinated even more tightly. These complexes are characteristically
thermodynamically stable when in solution. Polyazamacrocyclic complexes are more
stable than their non-cyclic analogs due to an increase in kinetic inertness, making them
of large interest in the medical applications field.4
The metal ion chosen to coordinate to the ligand will be dependent on the ring size of
the polyazamacrocycle and other donor atoms present.2 Two polyazamacrocycles in
particular that will be discussed further are 1,4,7,10-tetraazacyclododecane (cyclen), and
1,4,8,11-tetraazacyclotetradecane (cyclam). These two tetraazamacrocycles are of
particular interest due to the size of their cavity, the variety of synthetic routes available
to create the macrocycles, and the number of possible pendant arms that can be appended,
which will increase their possible functionality.2
1.01 Cyclam
Cyclam is a 14-membered ring in the tetraazamacrocyclic category, which is shown
in
Fig. 1. There are four nitrogen donor atoms that can be used to attach pendant arms
directly to the ring. Cyclam consists of two, 3-carbon bridges and two, 2-carbon bridges.
In more recent studies, cyclam has shown potential and is being further investigated as a
treatment for AIDs and for stem cell mobilization.5
When cyclam binds to a metal ion, typically 5- and 6-membered rings will be formed
that contain both the metal ion and the nitrogen donor atoms. Due to this, cyclam is more
suitable with slightly smaller metal ions opposed to very large metal ions. When pendant
arms are coordinated to the ligand, the metal ion becomes fixated in the center (cavity) of
the complex with additional ligation from the pendant arms. This allows for rigidity and
stabilization with chances of dissociation diminishing. Competing metal ions will be less
able to bind due to the stability of the newly formed compound.2
Cyclam has become the focus of new research focusing on MRI contrast agents, DNA
cleavers, luminescent probes, antigenic antibody labeling via radioisotopes, and in
radiopharmaceutical medicines.6 Cyclam shows promise in future research studies, with
emphasis as a mediator in HIV treatments due to its interactions with receptors.7
Figure. 1. The structure of 1,4,8,11-tetraazacyclotetradecane (cyclam)
.
1.02 Cyclen
Cyclen is a 12-membered ring, and like cyclam, contains four nitrogen donor atoms
as seen in
Fig 2. However, rather than alternating the number of carbons within the
bridges, cyclen contains only 2-carbon bridges throughout the structure. Like cyclam, the
donor atoms provide a location for pendant arms to be attached. Also like cyclam, cyclen
possesses the ability to form stable complexes with metal ions. Though cyclam is more
popular, cyclen also has been utilized in recent studies that examine the use of the
compound as an anti-tumor agent, in image contrast agents, and also in possible HIV
Figure 2. The structure of 1,4,7,10-tetraazacyclododecane (cyclen).
When compared to cyclam, cyclen is smaller by two carbons atoms. Cyclen has the
ability to bind a wide variety of metal ions, including ions of transitional metals.2
1.03 Cyclen Macrocycle in Medicinal Application
A useful derivative of cyclen is 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetate,
or DOTA. DOTA is currently used as MRI contrast agents known as Dotarem® and
ProHance® Further clinical studies are being conducted for the radiotherapeutic drug
chelator known as OctreoTher® incoorporating DOTA. In a 2008 study by Jiang
et al,
DOTA was examined as a potential 1H-19F dual nuclei MRI contrast agent along with
other potential macrocycles. NMR spectral characteristics were observed from the
complexes to determine T1 and T2 relaxation times that would be relevant in determining
if the macrocycles would be suitable as an MRI contrasting agent. From other research,
DOTA appears to have a large potential in this field, but clinical trials still need to be
conducted to ensure the methods are safe and effective.9
1.04 Importance of Macrocyclic Metal Ion Complexes in Medical Applications
It has been previously discussed that the lack of flexibility in some macrocyclic
ligands can be viewed as a positive side effect. Macrocyclic receptors are less flexible
than acyclic receptors, which means that they are pre-organized for binding to biological
targets. Knowing this, macrocycles are being studied intensely as possible drug delivery
Macrocycles continue to provide advantages in different biomedical applications over
some current techniques. Contrast agents in particular show advantages when
incorporating macrocyclic ligands. It is important to continue research in this area to
improve the methods that are currently available so that these agents are the most
effective and safest possible.12
Besides contrast agents, metal ion complexes are also of interest in anticancer agents
and possible Alzheimer's treatment.13,14 Currently there are 68 different macrocyclic
structures registered for drug administration. Of these, 10 are used for the treatment of
certain cancer types.12 However, when it comes to synthesizing a metal ion complex with
the intention of a medicinal application, it is necessary to be wary that toxic metals do
accumulate within the body. When considering which metal will be most stable and best
suited for the task, the degree of excretion needs to be considered.
Though there are a variety of possible metals to examine for use in a medicinal
application, cobalt is particularly well studied. There are ongoing studies examining
cobalt complexes as HIV protease inhibitors and as potential therapeutics for
hyperthermia. A clinical study is also being conducted on Doxovir®, which contains
cobalt, as a herpes simplex virus drug-resistant treatment.15 Cobalt(II) will later be
examined in this thesis as a potential coordinating metal ion in the case of MRI
contrasting agents.
1.05 Optimum Binding of Metal Ions
There are two major factors that ensure that a metal ion binds to a ligand in a strong
and stable manor--complementarity and constraint. When both factors are in place, the
complex will be satisfactorily stable.16
Factor 1: Complementarity—The complementarity of a complex refers to how well
the metal ion corresponds to the ligand. When the metal ion is in the cavity of the ligand,
it must be able to adapt to the properties of that ligand, such as the size and shape. The
metal ion will also share some electronic properties with that ligand. For the complex to
be most stable, these factors need to be optimal: electronic properties of the donor atoms,
how many donor atoms, bond length, and the overall structural arrangement.16 When
these factors are favorable, the complex will be very stable.
Factor 2: Constraint—Constraint is concerned with the overall flexibility of the
complex. Depending on the metal ion used and the bond lengths, overall flexibility will
vary. Constraint can be further broken down into two factors; topology and rigidity.
1.06 Topology
Topology relates to the 3-D spacing and overall geometry of a complex. When more
donor atoms are present, the binding constant will increase. This phenomenon can be
referred to as the chelate effect.16 Chelating ligands contain more than one donor atom
linked together. With more donor atoms linked together in the correct way, a higher
affinity towards a metal ion will be observed. Both cyclam and cyclen contain four
nitrogen donor atoms that can potentially bind to a metal ion, so these complexes will
possess more stability than those with a similar monodentate ligand.
Scheme 1.
With more individual molecules, there is more entropy than with fewer molecules,
thus creating stability (
Scheme 1).16, 17, 18 When a polydentate ligand such as cyclam
displaces four monodentate ligands, the formation constant is greater for the polydentate
ligand, thus producing a more stable complex than that with the four monodentate
Another theory behind the chelate effect involves the step-wise binding of the donor
atoms, shown in
Fig 3. In a monodentate ligand, the length of the bond between the metal
ion and second donor atom is dependent upon the donor atom's concentration. For a
polydentate ligand, the bond distance for the second donor atom is fixed due to the bond
with the first donor atom. Due to this, the rate at which the second donor atom of a
chelate binds to the metal ion is much quicker when compared to a monodentate donor. It
is also known that a donor atom of a polydentate ligand is able to dissociate at a rate
similar to a monodentate ligand. However, the rebinding of the second donor in a
polydentate is fast, thus creating a more kinetically stable complex when compared to
monodentate ligands.16
Figure 3. Binding of donor atoms in chelate versus monodentate ligands.19
It has been established that there is an increase in stability with polydentate,
macrocyclic ligands. The macrocyclic effect states that when a larger complex's donor
atoms are fixed together in a ring, a much sturdier complex is created.20 In typical acyclic
ligands, each donor atom can dissociate from the metal ion in an SN1-like mechanism.
However, when the atoms are linked together in a macrocycle. The SN1 mechanism
cannot occur successively as it would for an acyclic polydentate ligand. It is unlikely the
complex with a macrocyclic ligand would dissociate unless one bond became
significantly weaker and the complex distorts, causing a chain reaction of dissociation.18
1.07 Rigidity
Rigidity in chemical complexes pertains to the lack of flexibility of an array of donor
atoms. It has been previously found that macrocyclic ligands are less flexible than other
ligand types, but even within the scope of macrocycles, flexibility can vary. When donor
atoms are fixed in a specific orientation, the ligand structure will allow the metal ion to
bind in only a limited number of orientations. Due to a lack of movement, the ligand may
not be able to distort its shape to accommodate some metal ions. However, for metal ions
that can bind, the complex will be very stable. The complex is also now less able to
dissociate due to the fixed orientation of the structure.21 When the ligand is rigidified, the
corresponding complexes will ultimately be more stable.22
1.08 Pyrazoles
Pyrazoles are a group of compounds first synthesized in 1898 by Hans von Pechmann
from diazomethane and acetylene.23 Pyrazole is a heterocyclic, organic, 5-membered ring
compound. This 5-membered aromatic ring consists of three carbon atoms, two nitrogen
atoms, and two double bonds, which can be seen in
Fig 4. Substituted pyrazoles have a
plethora of uses in modern medicine. These compounds can be used as an analgesic as
well as an antipyretic, while other substituted pyrazoles can provide anti-inflammatory
properties.24 Pyrazole derivatives have been distributed in a variety of commercially
available drugs, including; Rimonabant®, Tefoxalin®, Isolan®, Lonazolac® and
Figure 4. The chemical structure of pyrazole.
There are many common derivatives of pyrazole available, including 3,5-
dimethylpyrazole, (
Fig 5). The dimethylated derivative exhibits greater anti-
inflammatory properties as compared to the un-substituted pyrazole, which exhibits
stronger analgesic and antipyretic characteristics.24
Figure 5. The chemical structure of 3,5-dimethylpyrazole
1.09 Pendant Arms
Coordinating pendant arms are used in macrocyclic chemistry so that more donor
atoms can potentially bind to a metal ion. Thus, when coordinating pendant arms are
used, they will also help fix the metal ion in the center of the ligand. Noncoordinating
pendant arms do not contain metal-binding groups, and therefore provide other
functionality.26
In the case of coordinated pendant arms, three functional groups are frequently
encountered: alkoxides, carboxylates, and amides.26 The characteristics of a metal ion
complex can be completely changed by such pendant groups, (such as the intermolecular
and intramolecular equilibria).26 The negative charge associated with one of these
functional groups will also help to reduce the overall charge of the complex due to the
presence of the cationic metal ion.
1.10 Coordination Number
Coordination numbers indicate how many donor atoms are attached to a central metal
ion.27 Tetraazamacrocyclic ligands with four pendant arms could have a coordination
number of up to eight. In this particular case, the macrocyclic ligand and the four
pyrazole pendant arms could be bonded to the central atoms in two limiting geometries—
square prismatic or anti-prismatic geometry. An example of prismatic versus anti-
prismatic coordination is shown in
Fig 6.
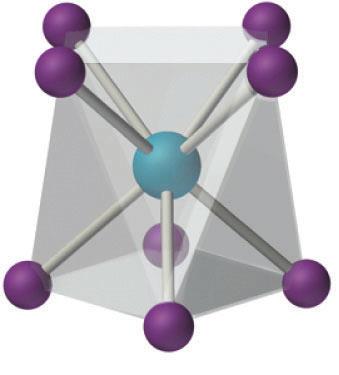
Figure 6. Square prismatic, left, versus antiprismatic, right, example.28,29
1.11 Magnetic Resonance Imaging
Paul Lauterbur first developed magnetic resonance imaging, or the MRI, in 1971.30
Though many other people are credited with attributing ideas that ultimately led to the
MRI instrument, John Mallard is credited with building the first MRI able to perform a
full body scan to detect and locate abnormalities and cancerous tissue in the late 1970's.30
An MRI works by placing the object to be imaged in a strong magnetic field that will
organize the spin states of each hydrogen atom present. These spin states will orient in
two possible ways, cancelling each other out for the most part. However, there will be a
small excess of protons in the lower energy state that will not be completely cancelled
and a net magnetic moment is generated in the direction of the field.31 The frequency of
radiation required to flip the magnetic moment vector depends on the strength of the
field. Finally, a field gradient is used and repeatedly turned on and off, resulting in a
variation of the magnetic field, which will allow for the "flipping" frequency to be varied
in three dimensions.32
The difference between the electromagnetic radiation emitted and absorbed by the
hydrogen nuclei in bodily water is what is measured in MRI. Ultimately, the MRI is
detecting differences in bodily water concentrations, which will exhibit contrast based on
composition (bone, tissue, etc). An image is then generated based on the data collected.
In Fig 7, an MRI image can be seen. 33
Figure 7. Generated MRI image
1.12 MRI Contrast Agents
Contrast agents are a useful tool in everyday medicinal imaging applications and not
isolated to MRI. The purpose of contrast agents is to generate a more intense image
making pathology more visible between normal tissue and diseased tissue. In the case of
MRI contrast agents, the objective is to enhance (or lessen) the polarization of the
hydrogen nuclei of the bodily water, thus leading to an enhanced (or decreased) signal
near the contrast agent. The instrumentation then observes and records the spin
polarization, and shortly after the protons will start to decay back to equilibrium. This is
due to what is known as a spin-lattice or T1 relaxation. T1 indicates how long it takes for
the proton spin states to return to thermal equilibrium, and will vary based on where in
the body the proton is located. Ultimately this difference can also be used to create
contrast. When a contrast agent has been introduced, the aim is often to decrease the T1 of
the protons being detected.34 With the proper pulse-sequence in the MRI, the result of this
will be an image with greater contrast, as noted in Fig 8.
Figure 8. MRI with no contrast, left, versus contrast, right.35
The difference between positive and negative contrast agents is due to their magnetic
properties. A positive agent can decrease T1 (longitudinal) rates, generating a large
difference between T1 and T2 relaxation. When the contrast image is generated via this
method, the regions of the image with more contrast agent will be brighter. Negative
contrast agents will tend to decrease spin-spin or T2 (transverse) rates, which will have
the effect of darkening the image in the area of the contrast agent. T2 relaxation causes
dephasing to occur, which is where the spins exchange energy with other spins in close
proximity. This differs from T1 where spins lose energy to the surroundings.36 MRI
contrast agents are for the most part positive, enhancing T1 relaxation. Though each type
possesses different characteristics, they each are important when diagnosing via MRI.34
Generally, T1 MRI contrasting agents are derived from gadolinium(III) complexes,
which is the preferred choice due to its strong paramagnetic nature.2,21 These types of
agents have proven to be effective, but may not always be the safest. Gadolinium agents
can bypass the blood brain barrier (BBB) and be nephrotoxic as well as neurotoxic. In
isolation, gadolinium can be toxic, which is definitely of concern. However, when the
element is localized within a chelated complex it is generally safe and a valuable resource
for the radiologist. When chelated, the gadolinium ion is held into the ligand tightly,
making it unlikely the element will escape and roam the body. Regardless, repeated
exposure to gadolinium can prove to have damaging effects, especially if the patient is
already suffering from a secondary health issue. 34
In 2006, studies provided evidence of a relationship between gadolinium (as a
contrast agent) and nephrogenic systemic fibrosis (NSF). Generally, patients who were at
highest risk for NSF had poor renal function. Patients were not being monitored for renal
function prior to being administered the gadolinium contrast agent, and therefore
suffering in the long run. It was also discovered that gadolinium was able to bypass the
BBB in patients with adequate renal function, and deposit in the endothelial walls of the
MRI is very useful in examining the central nervous system. The majority of
gadolinium-enhanced MRI scans are performed to observe the brain for abnormal
pathology. However, MRI can also be used to aid in identification of bodily
abnormalities in other areas such as the lymph nodes, heart, bone marrow, etc. There are
elements other than gadolinium that can be used to aid in enhancement of particular
tissues such as fluorine or manganese. Manganese is used in Manganese Enhanced
(MEMRI) MRI to detect heart disease. Manganese naturally has an attraction to the
myocardium, and this is exploited in this technique to help detect abnormality or
1.13 Gadolinium (III) Paramagnetic Relaxation
When a contrast agent is introduced the objective is to have accumulation of the agent
in tissues where abnormalities are observed. Areas that experience accumulated agent
will then also display enhanced T1 and/or T2 relaxation.38 Both the metal from the
contrast agent and the water nuclei in bodily tissue contain nuclear or electronic spins.
When dipole interactions then transpire between the two species, a phenomenon known
as Paramagnetic Relaxation Enhancement (PRE) is the result.39
Magnetism is a distinct property that relates to electrons. A complex with a metal ion
can be either diamagnetic or paramagnetic in nature. If diamagnetic, then the molecule
has only spin-paired electrons. If paramagnetic then the molecule will have at least one, if
not more, unpaired electrons present.40 Paramagnetism is observed when an external
magnetic field is present and objects are attracted to the field. This attraction takes place
due to the formation of induced magnetic fields that share the same direction as the
generated magnetic field. However, diamagnetism is the opposite where very weak
repulsion is the outcome. The material repels the magnetic field because the object's
small induced magnetic field is generated in the opposite direction as the actual applied
magnetic field.41 Gadolinium (III) is a paramagnetic ion.
Gadolinium(III) possesses a 4f electron shell with seven unpaired electrons. When
this metal ion binds to another molecule, these seven electrons maintain independence,
therefore keeping gadolinium a strongly paramagnetic ion. Gadolinium(III) also contains
up to nine sites for possible ligand binding to occur, but most ligands currently used for
contrast agents contain only eight donor atoms.39 A water molecule in the surrounding
area from solvent will then bind to the metal ion's ninth site. The water bound at this
ninth site will be in very close proximity to the metal. Once this final interaction takes
place, inner sphere relaxation of the water proton's nuclear spin will be triggered by the.
Inner sphere relaxation is a phase of paramagnetic relaxation enhancement where T1 and
T2 relaxation will occur together.39,42
1.14 ParaCEST
ParaCEST stands for "paramagnetic chemical exchange saturation transfer" as
described by Olatunde and colleagues.43 ParaCEST contrast agents possess
paramagnetism as well as the ability to exchange protons or water molecules with bulk
water.44 Currently, transition metals are typically used for paraCEST complexes due to
their paramagnetic nature and relative non-toxicity compared to gadolinium in clinical
situations. Ultimately, the aim is to use these complexes as alternate MRI contrast agents
due to their ability to be switched on and off depending on if a saturating pulse is
1.15 Magnetization Transfer Effect
The magnetization transfer effect can be utilized in MRI when the protons from the
body's water molecules and the exchangeable protons from the paraCEST agent resonate
at different frequencies. To obtain optimal results, the largest possible frequency
difference between the two protons (water molecule protons and paraCEST agent
protons) is desirable. This can be done through alterations to the ligand itself or trial runs
with alternate transition metals. If the changes made do prove successful and produce a
larger difference in frequency, then the complexes will exhibit selective contrast
enhancement and could prove to be highly important for the future of MRIs.7 The large
frequency differences between the exchangeable proton and bulk water protons will
ultimately make selective irradiation of the exchangeable protons an easier feat.43 An
example of a paraCEST spectrum of 1,4,7-tris(carbamoylmethyl)-1,4,7-
triazacyclononane is shown in Fig 9.
Figure 9. 1,4,7-tris(carbamoylmethyl)-1,4,7-triazacyclononane ParaCEST spectrum.46
T1 plays a significant role when it comes to paraCEST complexes. In previous
studies, Morrow and colleagues showed that compounds produced the highest resolution
CEST image when there was insignificant T1 water relaxation.35 This result shows that
the T1 water relaxation competes with the normal CEST pathway, and if the T1 relaxation
process is not operating strongly, then CEST is the primary cause of contrast in the MRI.7
1.16 Previously Synthesized ParaCEST Complexes
In 2013, a study performed by Dorazio et al. examined different macrocyclic ligands
to synthesize metal ion complexes with possible paraCEST applications. These
macrocycles included derivatives of 1,4,7-triazacyclononane (TACN), 1,4,7,10-
tetraazacyclododecane (cyclen), and 1,4,8,11-tetraazacyclotetradecane (cyclam).
Focusing on cyclen and cyclam, each has four sites for a coordinating metal ion to bond
to. In this particular study, a few different pendant groups were studied, including
pyridine derivatives. All were analyzed as pendant groups due to their ability to stabilize
transition metal ions holding a 2+ charge. Benzimidazole groups were also among the
choices due to their strength as a donor group. Finally, a TPT (1,4,7-tris(pyrazol-3-
ylmethyl)-1,4,7-triazacyclononane) ligand with three pyrazole pendant arms was
examined with Fe(II), Ni(II) and Zn(II).7
In the study it was noted that when a larger metal ion was used, the twist angle
between the nitrogens from the macrocycle ligand and nitrogens from the pendant
pyrazoles increased (square prismatic versus anti-prismatic structure). These coordination
geometries will ultimately effect the CEST properties of the structure. The synthesized
complexes proved to be stable and did not dissociate when in a neutral water at 37 oC.
From this study it was concluded that these complexes exhibited promise in future use as
ParaCEST agents but would need to undergo further in vivo testing to ensure that they are
safe and effective.7
1.17 Macrocyclic Ligands used as MRI Contrast Agents
Contrast agents have provided an advantage in imaging when it comes to pathology.
Macrocyclic ligands have proven to be desirable in the design of contrast agents and can
provide an advantage when used instead of an acyclic ligand.2 Compared to other ligands,
macrocycles generally provide more stability through formation and functionalization. In
a 2010 review, ligand design is discussed and how ultimately different designs can alter
stability of the overall complex.2 For instance, one particular study found that cyclam
ligands were less stable when complexed with lanthanides than were cyclen ligands.2
There has also been research performed on cyclen that maintains the size of the
macrocycle but increases the rigidity by incorporating a piperidine ring into the backbone
while also leaving four pendant arms in place. In this study, the synthesized ligand (seen
in Fig 10) was coordinated to gadolinium(III) and exhibited reduced kidney retention as
well as an increase in liver uptake when compared to the DOTA complexes traditionally
Figure 10. Rigidified ligand of 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic
acid (DOTA) adapted from Chong et al.47
In a 2008 study performed by Seeger et al, the differences between macrocyclic
contrast agent and linear contrast agent was examined.48 It was concluded that the
macrocyclic contrast agent (gadobutrol) required a lower dose for results comparable to
the linear contrast agent (gadopentetate dimeglumine). In conclusion, using a macrocyclic
contrast agents will allow for a lower administration dose, as well as decreasing the
possibility of dissociation of gadolinium.48
From the previously synthesized complexes, it is clear that the focus of contrast
agents used in MRI is shifting. Realizing the possibilities of contrast agents derived from
macrocycles coordinated to a biologically important metal ion that is stable in the
environment of the body, that has a high water proton transfer/exchange rate, and that can
be excreted more readily has led to our attempts to synthesize new macrocyclic ligands
with pyrazole pendant arms and their metal ion complexes.
2. Material and Methods
2.1. Materials
3,5-Dimethylpyrazole, pyrazole, 2-methoxyethanol, cyclam, cyclen hydrochloride,
1,2-dibromoethane, 1,2-dichloroethane, sodium bicarbonate, tetrabutylammonium
bromide, tetrabutylammonium hydrogen sulfate, sodium bicarbonate, diethyl ether,
methylene chloride, hydrochloric acid, ammonia hydroxide, anhydrous sodium sulfate,
anhydrous magnesium sulfate, sodium hydroxide, copper acetate, methanol,
dimethylformamide, iron(II) tetrafluoroborate hexahydrate, tetrahydrofuran, copper(II)
perchlorate hexahydrate, cobalt(II) chloride hexahydrate, and acetonitrile were obtained
from commercial sources and used as received.
The solvents acetonitrile, DMF, and THF were dried prior to use.
2.2. Methods
General: All reactions were run under nitrogen or argon with stirring.
Instrumentation: High resolution MS data was collected for all ligands as dilute
samples in methanol on a Thermo Scientific Exactive with an electrospray source.
Samples were introduced at a flow rate of 5 μL per minute using an infusion pump. For
fragmented MS, fragmentation was performed using 30 kV fragmentation energy.
All 1H and 13C NMR data was obtained using a Bruker Avance III 400 MHz NMR.
Tetramethylsilane (TMS) was used as an internal standard and all samples were recorded
in CDCl3 (deuterated chloroform) at 300 K.
The IR data collected for each of the four ligands was recorded using diamond
attenuated total reflectance (ATR). Data collection was performed on a Thermo Scientific
Nicolet iS50 FT-IR.
X-ray crystallography was performed on all four ligands using a Bruker SMART
APEX II CCD or a Bruker D8 QUEST diffractometer. Crystals obtained were adhered to
a 0.1 mm diameter Mitigen MicroLoop at -100 oC. MoKα radiation was used to collect
data. Based on noted absences and statistical intensity, space groups were then
determined. To examine remnant non-hydrogen atoms, Fourier cycles were performed.
Hydrogen atoms were then able to be located and positioned.49,50,51
1-(2-Bromoethyl)pyrazole (BP). In a 300-mL roundbottom flask, 4.0 g (0.059 mol)
of pyrazole, 4.0 g (0.088 mol) NaOH pellets, 12 mL water, 170 mL (2.08 mol) of 1,2-
dibromoethane and 0.70 g (0.0022 mol) tetrabutylammonium bromide were heated to
reflux for 16 hours. The reaction was allowed to cool, and a liquid-liquid extraction was
performed with diethyl ether and aqueous sodium bicarbonate. The organic layer was
dried with anhydrous sodium sulfate and then evaporated under vacuum to dryness. The
1H NMR spectrum was identical to that reported by López et al.52
1-(2-Chloroethyl)pyrazole (CP). In a 25-mL roundbottom flask, 2.0 g (0.029 mol)
of pyrazole, 14.6 g (0.0146 mol), 10.0 mL of 1,2-dichloroethane, 1.8 g (0.044 mol) of
solid NaOH pellets, 4 mL of water, and 0.35 g (0.0011 mol) of tetrabutylammonium
hydrogen sulfate were heated to reflux for two hours and allowed to cool. A liquid-liquid
extraction was conducted with aqueous sodium bicarbonate and diethyl ether. The
organic layer was dried with anhydrous sodium sulfate, and evaporated in vacuo. The 1H
NMR spectrum was identical to that reported by Attarian et al.53 All other
characterization was also consistent with previous literature values.
1-(2-Chloroethyl)-3,5-dimethylpyrazole (DiMeCP). In a 100-mL roundbottom
flask, 3.3 g (0.083 mol) NaOH, 4 mL of water, 4.0 g (0.042 mol) of 3,5-
dimethylpyrazole, 58 mL (0.73 mol) of 1,2-dichloroethane, and 0.50 g (0.0015 mol)
tetrabutylammonium hydrogen sulfate were heated to reflux for 16 hours. The reaction
was cooled and extraction was performed with diethyl ether and aqueous sodium
bicarbonate. The organic layer was dried with anhydrous sodium sulfate, and dried in
vacuo. The 1H NMR spectrum was identical to that characterized by Attarian et al.53 All
other characterization was also consistent with literature values.
1-(2-Bromoethyl)-3,5-dimethylpyrazole (DiMeBP). In a 300-mL roundbottom, 3.3
g (0.034 mol) of 3,5-dimethylpyrazole, 3.0 g (0.078 mol) solid NaOH, 10 mL of water,
140 mL (1.7 mol) dibromoethane and 0.58 g (0.0017 mol) of tetrabutylammonium
bromide were heated to reflux for 16 hours. After cooling, a liquid-liquid extraction was
conducted using diethyl ether and aqueous sodium bicarbonate. The organic layer was
dried with anhydrous sodium sulfate and evaporated in vacuo. The 1H NMR spectrum
was identical to that reported by Lopez et al. All other characterization was also
consistent with literature values.52
Tetraethylpyrazolyl cyclam (PzCm). In a 10-mL roundbottom, 50 mg (0.00025
mol) of cyclam, 195 mg (0.0015 mol) of CP, 2 mL (0.025 mol) 2-methoxyethanol, and
400 mg (0.0047 mol) of NaHCO3 was heated to reflux for 72 hours. A liquid-liquid
extraction was performed using aqueous sodium bicarbonate and methylene chloride. The
organic layer was dried with anhydrous magnesium sulfate, and evaporated in vacuo. The
crude product was then dissolved in water/methanol, from which the product crystallized
out as white crystals with yield of 0.0322 g (4.4%). 1H NMR δ (ppm) = 1.44 (m, 4H, J =
6.8 Hz); 2.41 (m, 16H suspected overlap); 2.81 (t, 8H, J = 6.6 Hz); 4.12 (t, 8H, J = 6.4
Hz); 6.20 (t, 4H, J = 2.1 Hz), 7.40 (d, 4H, J = 2.2 Hz), 7.48 (d, 4H, J = 1.8 Hz). 13C NMR
δ (ppm) = 24.0, 50.5, 51.7, 55.0, 106.0, 129.7, 139.2. HRMS calculated for C30H48N12 +
H+ m/z = 577.4198, actual m/z = 577.4197. IR: 2949, 2796, 1513, 1455, 1395, 1353, 1311
Tetraethylpyrazolyl cyclen (PzCn). In a 25-mL roundbottom flask, 400 mg (0.0023
mol) of cyclen tetrahydrochloride, 1.1 g (0.0084 mol) of BP, 1.0 g (0.012 mol) of
NaHCO3 and 16 mL of 2-methoxyethanol were heated to reflux for 24 hours. An
additional 1.0 mL of BP was added at this time, and the heating was continued for
another 24 hours. The reaction was cooled and an extraction was then performed. First,
an extraction into acid (pH 4) from methylene chloride was performed. Three washes
with methylene chloride were performed to extract any residual organic. The acid layer
was then made basic (pH>10) with concentrated ammonia hydroxide. Three extractions
with methylene chloride were then performed. The organic layer was dried with
anhydrous sodium sulfate and evaporated in vacuo. The resulting solid was dissolved in a
small amount of methylene chloride and diethyl ether and crystallized, resulting in 0.183
g (16%) of colorless crystals. 1H NMR δ (ppm) = 2.42 (s, 16H); 2.76 (t, 8H, J = 6.3 Hz);
4.07 (t, 8H, J = 6.3 Hz); 6.17 (t, 4H, J = 2.1 Hz), 7.41 (d, 4H, J = 2.2 Hz), 7.47 (d, 4H, I =
1.8 Hz). 13C NMR δ (ppm) = 50.4, 53.8, 55.9, 105.3, 130.2, 139.4. HRMS calculated for
C28H44N12 + H+ m/z = 549.3885, actual m/z = 549.3889. IR: 3105, 2810, 2944, 1516,
1447, 1396, 1372, 1356 cm-1.
Tetra-3,5-dimethylpyrazole cyclam (DiMePzCm). In a 50-mL roundbottom, 330
mg (0.00160 mol) of cyclam, 1.3 g (0.0090 mol) of DiMeCP, 2.7 g (0.32 mol) of solid
sodium bicarbonate, and 20 mL of 2-methoxyethanol was heated to reflux for three days,
72 hours. The reaction was cooled and an extraction was conducted using diethyl ether
and aqueous sodium bicarbonate. The organic layer was dried with anhydrous sodium
sulfate, and evaporated in vacuo. The resulting solid was dissolved in water and methanol
and allowed to crystallize, yielding 0.558 g (49%) of colorless crystals. 1H NMR δ (ppm)
= 1.52 (m, 4H, J = 6.8 Hz); 2.19 (s, 12H); 2.22 (s, 12H); 2.49 (m, 16H); 2.76 (t, 8H, J =
7.0 Hz); 3.95 (t, 8H, J = 7.0 Hz); 5.74 (s, 4H). 13C NMR δ (ppm) = 11.5, 13.8, 24.5 47.4,
52.3, 55.5, 105.1, 139.1, 147.6. HRMS calculated for C38H64N12 + H+ m/z = 689.5450,
actual m/z = 689.5453. IR: 2929, 2810, 1550, 1457, 1422, 1383, 1369 cm-1.
Tetra-3,5-dimethylpyrazole cyclen (DiMePzCn). In a 25-mL roundbottom, 400 mg
(0.00230 mol) of cyclen, 1.0 g (0.012 mol) of NaHCO3, 16 mL of 2-methoxyethanol, and
1.2 g (0.0085 mol) of DiMeCP was heated to reflux over 24 hours. The reaction was
cooled and a liquid-liquid extraction was performed with methylene chloride and basic
water (ammonia hydroxide). The organic layer was dried with anhydrous sodium sulfate
and evaporated under vacuum. The product was not pure enough to crystallize, so a
column on silica was performed. Pure fractions were collected and subjected to another
liquid-liquid extraction with methylene chloride and basic water (ammonia hydroxide).
The organic layer was then dried with sodium sulfate and dried under vacuum overnight.
To crystallize the product, the solid was dissolved in ether (50 mL) with a few drops of
methanol. Crystals were long, thin, and colorless and ultimately yielded 0.114 g (14%).
1H NMR δ (ppm) = 2.18 (s, 12H); 2.22 (s, 12H); 2.53 (s, 16H); 2.73 (t, 8H, J = 7.0 Hz);
3.95 (t, 8H, J = 7.2 Hz); 5.73 (s, 4H). 13C NMR δ (ppm) = 11.2, 13.5, 46.7, 53.4, 55.6,
104.8, 138.8, 147.2. HRMS calculated for C36H60N12 + H+ m/z = 661.5137 and actual m/z
= 661.5135. IR: 2955, 2935, 2814, 1551, 1464, 1422, 1377 cm-1.
Metal Ion Complexation Studies.
Copper Acetate in Methoxyethanol. In a 5-mL roundbottom, 50 mg (0.000073 mol) of
DiMePzCm, 10 mg (0.00005 mol) of copper acetate, 1.0 mL of 2-methoxyethanol and
1.0 mL (0.025 mol) of MeOH was added and heated to reflux for one hour. Upon
addition of ligand and metal-containing compound an immediate color change was noted
from blue to green. The roundbottom was then sealed and left in the freezer over the
course of a night. No crystals had formed so the liquid was evaporated and replaced with
diethyl ether. It was then placed back in the freezer. However, no crystals formed.
Copper Acetate in Dimethylformamide. In a 10-mL roundbottom, 50 mg (0.000076
mol) of PzCn, 28 mg (0.00014 mol) of copper acetate, and 5 mL of DMF were added
together and heated to reflux over a five day period. The solution was a bright blue color.
The liquid was evaporated and acetonitrile was added to attempt crystallization. No
complexes or crystals formed. A blue substance left over in the flask and believed to be
unreacted copper acetate, perhaps due to ligand decomposition.
Iron (II) tetrafluoroborate hexahydrate THF. In a 5-mL roundbottom, 50 mg
(0.000086 mol) of PzCm, 25 mg (0.000074 mol) iron (II) tetrafluoroborate hexahydrate,
and 1.0 mL THF were added and heated to reflux over a one hour period. A color change
was noted to a dark brown. The roundbottom was then sealed to prevent evaporation and
placed in the freezer to facilitate crystallization. No crystals were formed.
Copper perchlorate hexahydrate in methanol. In a 5-mL roundbottom, 50 mg
(0.000073 mol) DiMePzCm, 29 mg (0.000078 mol) of copper perchlorate hexahydrate,
and 1.0 mL of MeOH were added and heated to reflux for one hour. When the ligand was
added to the copper, a color change occurred immediately from blue to green. After
heating, the roundbottom was sealed and placed in the fridge for 4 hours. No crystals had
formed so liquid was evaporated and replaced with diethyl ether and put into the freezer.
Again, no crystals formed.
Chapter 3. Results and Discussion
3.1 Pyrazoles as Pendant Arms
The pyrazole groups (either 3,5-dimethyl or unmethylated) were synthesized with
either a bromine or chlorine substituent. Dichloroethane and dibromoethane were used to
create the two carbon bridge used to connect the pyrazole arm to the ligand. 1-(2-
Bromoethyl)pyrazole and 1-(2-bromoethyl)-3,5-dimethylpyrazole are shown in Fig 11,
and 1-(2-chloroethyl)pyrazole and 1-(2-chloroethyl)-3,5-dimethylpyrazole can be seen in
Fig 12. Bromide and chloride are both excellent leaving groups that are advantageous
when it comes to bonding pendant arms to a ligand.
Figure 11. Chemical structure of 1-(2-bromoethyl)pyrazole (a) and 1-(2-bromoethyl)-
3,5-dimethylpyrazole (b)
Figure 12. Chemical structures of 1-(2-chloroethyl)pyrazole (a) and 1-(2-
chloroethyl)-3,5-dimethylpyrazole (b)
3.2 Tetraethylpyrazolyl Cyclam (PzCm)
Tetraethylpyrazolyl cyclam (PzCm) is a 14-membered ring with four pyrazole
pendant arms attached at each nitrogen atom of the macrocyclic ring, pictured in Fig 13.
In Scheme 2 the synthesis of the PzCm ligand is shown. When it came to synthesizing
the ligands, several solvents, bases, and reaction conditions were tried over the course of
several months of effort. Lutidine, acetonitrile, dimethylformamide, toluene, and several
other solvents were all used in hope of synthesizing the final ligands. Some reactions
were heated to reflux while others were stirred at room temperature to observe if this
could be a factor in yielding the wanted product. Unfortunately, although some product
was obtained in many cases, none of the solvents and conditions proved overly
successful. Ultimately, hot 2-methoxyethanol with solid sodium bicarbonate were the
reaction conditions settled upon for all reactions.
Figure 13. The structure of PzCm.
Scheme 2.
A high resolution mass spectrum was examined of this product to ensure that it was in
fact the desired product. The spectrum is exhibited in Fig 14. The bottom spectrum
displays the expected mass spectrum based on the chemical formula (C30H49N12). On the
top is a spectrum of the actual sample that was tested. The two spectra are almost exact.
The first peak at 577.4197 varies between the sample and expected by 0.0001 amu, or by
0.17 ppm. The second peak varies by 0.0002 amu. Though there is a slight discrepancy in
two of the peaks, the difference is 0.35 ppm and therefore is considered to be close
enough to be definitive. As the mass to charge ratio is almost exact between the actual
and expected, it can be concluded that the actual product is the same as the expected
tetramethylpyrazole
0.09-0.30 AV: 17 T: FTMS + p ESI Full ms
[120.00-2000.00]
580.4274 581.4203 582.6236
p (gss, s /p:40) Chrg 1
579.4264 580.4299 581.4333 582.4367
Figure 14. High resolution mass spectrum of PzCm actual (top) and theoretical (bottom).
To examine molecular structure, NMR is regularly used. In this case, both 1H and 13C
spectra were recorded to verify the final product was the expected product. The 13C NMR
in Fig 15 shows the expected number of carbons in the product, and based on their shifts,
it can be determined that this spectrum matches the expected compound. Fig 17 shows
the 1H NMR for the same PzCm crystals. A labeled structure of PzCm is also given to
compare to the NMR spectra (Fig 16). 2-D NMR (Appendix A) was used to help assign
the protons and the carbons. In this way it was possible to see the carbon at 24 ppm is
correlated to the hydrogen at 1.5 ppm. The carbon at 51 ppm is correlated to the 1H triplet
at 4.15 ppm. At 52 ppm, this carbon pairs with the hydrogen at 2.45 ppm. Carbon at 55
ppm pairs with the hydrogen triplet at 2.8 ppm. Finally, the carbon at 105 ppm pairs with
the hydrogen at 6.25 ppm.
Figure 15. 13C NMR of PzCm
Figure 16. Structure of PzCm for NMR assignment.
Figure 17. The 1H NMR spectra of PzCm
IR, or infrared spectroscopy, can also be used to identify functional groups. It is a
characterization method used to aid in the verification of final products. The IR of PzCm
spectra can be examined in Fig 18. There are only two peaks displayed that can be
readily assigned. At 2796 cm-1 and 2949 cm-1 there are short, broad peaks adjacent to
each other. The peak at 2796 cm-1 is indicative of an alkane C–H bond, as is peak at 2949
cm-1. There is also a small peak noted by arrow greater than 3000 cm-1, which is likely
due to the pyrazole-sp2 C-H. Peaks presented around 1500 cm-1 are likely due to carbon-
carbon and carbon-nitrogen bonds of pyrazole. However, the information collected does
not definitively conclude whether the synthesized product is the expected result or not
since there are not expected to be characteristic IR peaks in PzCm.
Figure 18. IR spectra of PzCm
The structure in Fig. 19 was determined by X-ray crystallography. It is evident that
the product in question is the product desired. The cyclam macrocycle can be observed
along with the four pyrazole pendant arms (unmethylated). The molecule sits on an
inversion center.
Figure 19. X-ray crystallographic structure of PzCm. Hydrogen atoms are omitted for
clarity.
The X-ray structure provides detail that other characterization data could not, such as
bond lengths, angles, and conformation. (Crystallographic information is outlined in
Table 1, bond lengths are detailed in Table 2 and bond angles in Table 3.) The blue
atoms in Fig 19 represent the nitrogens. In the orientation of the structure, the pyrazole
arms labeled 1 and 3 are extended in approximately the same plane, as approximately
defined by the four nitrogens of the macrocycle, as the macrocycle while pyrazole arms 2
and 4 are folded in, placed out of plane to the macrocycle.
Table 1. PzCm Crystallographic Information
Cell Lengths (Å)
a. 8.2513(7) b. 9.3828(7) c. 11.6523(9)
α. 109.012(2) β. 95.996(2) γ. 101.983(2)
Cell volume (Å3)
Table 2. PzCm Bond Lengths
Table 3. Selected bond angles from PzCm
3.3 Tetraethylpyrazolyl Cyclen (PzCn)
Cyclen is a 12-membered ring that has a similar structure to cyclam but has two-
carbon spacers between the nitrogens rather than alternating two- and three-carbon
spacers. Like cyclam, the nitrogen atoms of the cyclen ring constitute donor atoms that
the pyrazole pendant arms can also be bonded to. The structure of tetraethylpyrazole
cyclen can be observed in Fig 20. Scheme 3 displays the synthesis of PzCn.
Figure 20. The structure of PzCn.
Scheme 3.
The purified product was characterized by 1H and 13C NMR. Fig 21 shows the 1H
NMR and Fig 23 displays the 13C NMR. To better visualize which protons and carbons
are associated with the molecule, a labeled PzCn structure is presented in Fig 22. A
heteronuclear 2-D NMR (Appendix B) was performed to show C-H connectivity. From
this analysis, it was concluded that the carbon at 30 ppm is bonded to the hydrogen at 2.1
ppm, the carbon at 50 ppm is associated with the hydrogen at 4 ppm, the carbon at 54
ppm with the hydrogen at 2.5 ppm, the carbon at 56 ppm associated with the hydrogen at
2.8 ppm, and the carbon at 108 ppm is bonded to the hydrogen 6.2 ppm. The NMR
spectra is consistent with the structure.
Figure 21. The 1H NMR of PzCn.
Figure 22. The structure of PzCn labeled for NMR assignment.
Figure 23. The 13C NMR spectra of PzCn.
A high resolution mass spectrum was also recorded to observe the exact mass of the
compound (Fig 24). The main peak at m/z = 549.3885 is quite comparable to the
expected peak of 549.3889, differing by only 0.73 ppm. Mass spectral peaks were also
observed for the sodium ion adduct, in which there was also a good match with the
azole , indicating sodium
nt during ionization (Fig 24).
cyclen pyrazole#12
RT: 0.17 AV: 1 T:
FTMS + p ESI Full ms [120.00-1000.00]
Figure 24. High resolution mass spectrum of PzCn actual (top) versus theoretical
(middle) and with a sodium adduct theoretical (bottom).
An IR spectrum was also examined for functional groups to compare to the expected
product. Sharp peaks were observed at 2810 cm-1, 2944 cm-1, and 3105 cm-1. The peak
that appears at 2810 cm-1 is believed to be an sp3 C–H, though typically these peaks occur
between 2850-3000 cm-1. The peak at 2944 cm-1 also seems to belong to an sp3 C–H. As
for the peak at 3105 cm-1, this peak is the result of the pyrazole-sp2 C-H. Peaks present
around 1500 cm-1 are likely due to the carbon-carbon and carbon-nitrogen bonds within
the pyrazoles. The IR spectrum can be observed in Fig 25. However, the IR does not
provide any definitive conclusions to be drawn since the peaks belong to C–H bonds
cannot help positively identify the product in this instance.
Figure 25. IR spectrum of PzCn.
X-ray crystallography was used to examine the crystal structure of PzCn. The
structure can be seen in Fig 26. The cyclen backbone can be seen with four pyrazole
pendant arms bound to the nitrogen atoms of the macrocycle. This molecule is again
achiral and sits on an inversion center. Like the structure of PzCm, PzCn structure also
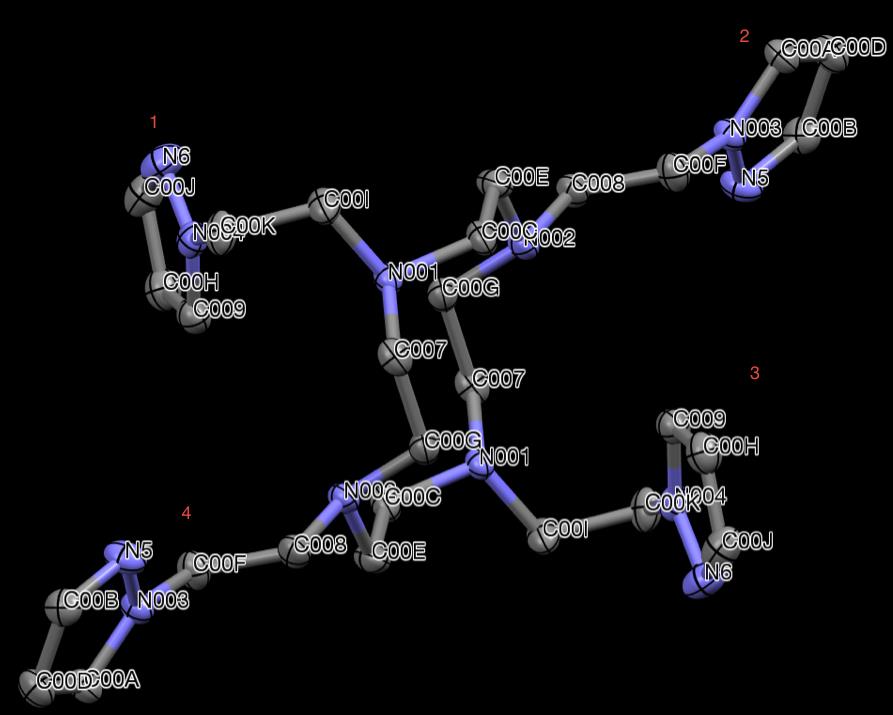
presents two pyrazole arms, 1 and 3, that are bent out of the (approximate) plane of the
macrocycle, while pyrazole arms 2 and 4 and stretched out within the plane defined
loosely defined by the macrocycle.
Figure 26. X-ray structure of PzCn. Hydrogen atoms are omitted for clarity.
The X-ray structure of PzCn provides minute details that are not obtainable
otherwise. Crystallographic data is tabulated in Table 4, bond lengths for PzCn are
detailed in Table 5, and selected bond angles in Table 6. From this information it was
determined all bond lengths were as expected and no solvent was present.
Table 4. PzCn Crystallographic Information
Cell Lengths (Å)
a. 9.1697(6) b. 9.2989(6) c. 9.6114(7)
α. 111.942(2) β. 96.475(2) γ. 101.075(2)
Cell volume (Å3)
Table 5. Bond lengths of PzCn.
Atom1 Atom2 Length N001
Table 6. Selected bond angles for PzCn.
3.4 Tetradimethylpyrazolyl Cyclam (DiMePzCm)
A dimethylated analogue of PzCm was also synthesized and examined. The structure
of DiMePzCm is the same as PzCm with the exception of the methyl groups on carbons
3 and 5 in the pyrazole rings of the pendant arms. It is expected that the macrocyclic
ligand should remain relatively unchanged by this addition. The chemical structure of
DiMePzCm can be seen in Fig 27. The synthesis of this molecule is displayed in Scheme
Figure 27. The structure of DiMePzCm.
Scheme 4.
NMR spectra were obtained for this compound for both 13C and 1H nuclei. The proton
spectrum exhibited in Fig 28 and a 13C spectrum is shown in Fig 30. A labeled structure
of DiMePzCm (Fig 29) shows the spectral peak assignments. 2-D NMR (Appendix C)
analysis was used to help identify which carbon correlated to which hydrogen. In
summary, the NMR spectra provided evidence that the expected groups were all present.
Figure 28. The 1H spectrum of DiMePzCm.
Figure 29. Labeled structure of DiMePzCm for NMR assignment.
Figure 30. 13C NMR spectrum of DiMePzCm.
After acquiring the NMR spectra, a high resolution mass spectrum was acquired to
ensure proper mass to charge ratio of the desired product. Fig 31 displays the mass
spectrum that was analyzed showing a peak at m/z = 689.5450, which is quite comparable
to the expected peak of 689.5453 only off by 0.44 ppm, indicating again that the product
is the expected one.
cyclam methylpyrazole#9-20
RT: 0.13-0.28 AV: 12 T: FTMS + p ESI Full
ms [120.00-1500.00]
p (gss, s /p:40) Chrg 1
Figure 31. High resolution mass spectrum of DiMePzCm actual (top) versus theoretical
(bottom).
Lastly, and IR spectrum was analyzed for functional groups present in the final
crystallized structure and is displayed in Fig 32. The IR exhibited two identifiable peaks
at 2810 cm-1 and 2929 cm-1. At 2810 cm-1, this peak is indicative of an sp3 C–H,
although again it falls slightly out of the typical range of 2850-3000 cm-1. The peak that
appears at 2929 cm-1 is also due to an sp3 C–H bond. Since this is the dimethylated
analogue, the C–H (pyrazole) is barely discernable. Again, this spectrum does not
provide any definitive data as the peaks represent C–H bonds, which cannot conclude
whether the product is or is not the expected product.
Figure 32. IR spectrum of DiMePzCm.
A structure of DiMePzCm was obtained by single crystal X-ray crystallography as
shown in Fig 33. The crystal structure displays the four 3,5-dimethylpyrazole groups and
the cyclam macrocycle confirming that the expected structure and the actual structure are
one in the same. The molecule is achiral and has a center of symmetry.
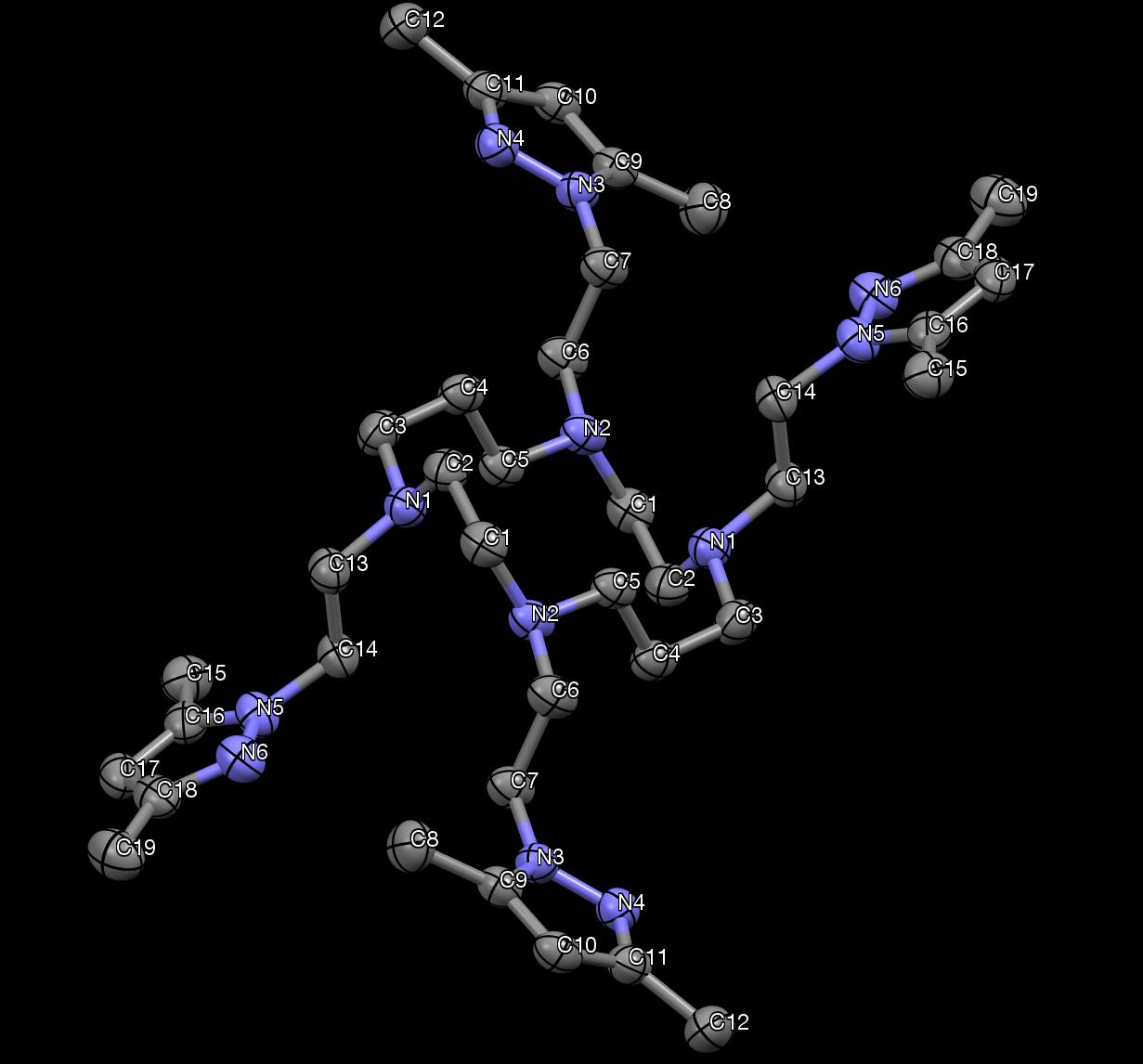
Figure 33. The X-ray crystallography of DiMePzCm. Hydrogen atoms are not shown.
From the X-ray structure of DiMePzCm, the inversion symmetry is evident.
Crystallographic data is provided in Table 7, bond lengths are provided in Table 8, and
selected bond angles in Table 9.
Table 7. DiMePzCm Crystallographic Information
Cell Lengths (Å)
a. 7.9021(4) b. 11.2591(6) c. 12.7366(9)
α. 63.853(2) β. 84.631(3) γ. 83.556(2)
Cell volume (Å3)
Table 8. Bond lengths of DiMePzCm.
Table 9. Selected bond angles of DiMePzCm.
3.5 Tetradimethylpyrazolyl Cyclen (DiMePzCn).
The final compound in the series is the cyclen with dimethylpyrazole pendant arms. It
is similar to the PzCn structure previously discussed, but this compound contains two
methyl groups on carbon 3 and 5 of each pyrazole. The macrocyclic ring remains
unaltered. The chemical structure can be viewed in Fig 34. The synthesis scheme can also
be viewed in Scheme 5.
Figure 34. Structure of DiMePzCn.
Scheme 5.
The purification of this compound differed slightly from the others. Ammonia
hydroxide was used as the base for the extraction. In an earlier attempt to extract the
product, the basic layer consisted of sodium bicarbonate, which in this case provided a
product that was clearly still partly partially protonated (NMR). The more basic ammonia
hydroxide work-up provided much more favorable results. Once a solid was recovered
from the organic layer and dried, it was dissolved into a large volume of ether,
approximately 50 mL. The solid was reluctant to dissolve, so a few drops of methanol
were added to facilitate dissolution of the solid. Once all was dissolved, the roundbottom
was refrigerated for a time span of 7 days. Long, flat, thin and clear crystals had
appeared. These crystals were then vacuum filtered and maintained for further
characteristic analysis via IR, mass spec, and carbon/proton NMR.
Protons and carbon NMR were recorded. A 1H NMR spectrum in Fig 35 was used to
compare with Fig 37. A 2-D spectrum was also recorded to aid in proton and carbon
assignments. A labeled structure of DiMePzCn (Fig 36) is shown with peaks assigned
with the help of 2D NMR (Appendix D).
Figure 35. The 1H NMR of DiMePzCn.
Figure 36. The labeled structure of DiMePzCn showing NMR assignments.
Figure 37. 13C NMR spectrum of DiMePzCn.
After ensuring the NMR spectra were consistent with the expected product, a high
resolution mass spectrum was obtained to compare the mass to charge ratio between the
theoretical and actual product. Fig 38 shows the fragmented high resolution mass
spectrum for the product (top) versus the expected mass spectrum based on the chemical
formula (bottom).
Examination of the HR MS with fragmentation energy applied revealed other details.
Peaks at m/z = 331.2604, 453.2450, and 661.5145 were observed. The peak at m/z =
331.2604 is due to the fact that sample was becoming doubly charged when introduced to
the MS. When the sample is doubly charged, the m/z is now halved. The isotopes of this
peak now also appear at 0.5 ppm apart rather than 1.0 ppm. The close up obtained (Fig
40) displays the sample which was suspected to be doubly charged versus the theoretical
with a charge of 2+. The actual differs from the theoretical by 1.2 ppm.
The molecular ion (with no fragmentation energy applied) can be examined in Fig 39.
The top spectrum belongs to the product while the bottom spectrum belongs to the
expected product based on the chemical formula. A peak evolved at m/z = 661.5135 for
the product where the expected peak displayed at m/z = 661.5137, which differed by only
0.30 ppm. This shows that the actual product is the expected product. But due to the
fragmented MS it can be concluded that this ligand in particular is not as stable due to the
dissociation of pyrazole pendant arms.
1.78E8cyclen tetradimepyrazole
frag#16 RT: 0.23 AV: 1 T:
FTMS + p ESI Full ms2
[email protected]
[120.00-2000.00]
1012.5753 1309.3871
Figure 38. High resolution mass spectrum of DiMePzCn fragmented actual (top) versus
theoretical (bottom).
tetradimepyrazole#1
RT: 0.02 AV: 1 T:
FTMS + p ESI Full ms
[120.00-2000.00]
Figure 39. Molecular ion mass spectrum of DiMePzCn.
tetradimepyrazole#1 RT: 0.02 AV: 1 T:
FTMS + p ESI Full ms
[120.00-2000.00]
C 36 H 62 N 12p (gss, s /p:40) Chrg 2
R: 0.001 Da @FWHM
332.2639 332.7656 333.2672 333.7689 334.2705
Figure 40. High resolution mass spectrum of DiMePzCn doubly charged.
An IR spectrum was obtained to complete the characterization, shown in Fig 41.
Three peaks presented, as can be found in Fig 25. These peaks appear at 2814 cm-1, 2935
cm-1, and 2955 cm-1. All three of these peaks are consistent with sp3 C–H groups. As
mentioned previously, the peak due to the pyrazole-sp2 C-H is barely discernable due to
this being the dimethylated analogue. Again, the IR cannot be used to definitely prove
whether the product is or is not the expected product to its results only providing data of a
Figure 41. IR spectrum of DiMePzCn.
X-ray crystallography was used to obtain a structure of the crystals produced. In Fig
42, all four dimethylpyrazole pendant arms can be identified as well as the cyclen
macrocycle. This structure allows conformation that the compound synthesized was that
expected. This compound is also achiral, seen with an inversion center. This complex is
also in the triclinic PĪ space group, as before.
Figure 42. X-ray crystal structure of DiMePzCn. Hydrogens are omitted for clarity.
From Fig 42 it can be observed that the structure contains inversion symmetry.
Crystallographic data is detailed in Table 10, bond lengths are detailed in Table 11 and
selected bond angles are tabulated in Table 12.
Table 10. DiMePzCn Crystallographic Information
Cell Lengths (Å)
a. 7.7806(6) b. 11.7098(9) c. 12.2792(9)
α. 105.243(3) β. 107.823(3) γ. 104.946(3)
Cell volume(Å3)
Table 11. Bond lengths of DiMePzCn.
Atom1 Atom2 Length C1
Table 12. Selected bond angles of DiMePzCn.
3.6 Comparison of Ligands
All four macrocyclic ligands were successfully synthesized by approximately the
same method, and each was crystallized over the course of the project. Through trial and
error, it was determined which method worked the most efficiently and in a timely
To make comparison easier, the data for MS, IR, and NMR can be observed in Table
13. Table 13 includes all characterization data for PzCm, PzCn, DiMePzCm, and
DiMePzCn.
Table 13. Characterization data for PzCm, PzCn, DiMePzCm, and DiMePzCn.
Compound
IR (cm-1)
(, ppm)
(, ppm)
1.44(m), 2.41(m),
24.0, 50.5, 51.7,
1311, 1353, 1395,
2.81(t), 4.12(t),
51.8, 55.0, 106.0,
1455, 1513, 2796,
6.20(t), 7.40(d),
2.42(s), 2.76(t),
50.4, 53.8, 55.9,
1356, 1372, 1396,
4.07(t), 6.17(t),
1447, 1516, 2944,
7.41 (d), 7.47(d)
1.52(m), 2.19(s),
11.5, 13.8, 24.5,
1369, 1383, 1422,
2.22(s), 2.49(m),
47.4, 52.3, 55.5,
1457, 1550, 2810,
2.76(t), 3.95(t),
2.18(s), 2.22(s),
11.2, 13.5, 46.7,
1377, 1422 1464,
2.53(s), 2.73(t),
53.4, 55.6, 104.8,
1551, 2814, 2935,
3.95(t), 5.73(s)
From Table 13, it is concluded that all previously discussed data is congruent with
the expected results for the ligands synthesized. The spectra are consistent with each
other, showing very similar chemical shifts between compounds of similar make-up.
Crystal structures were also obtained and are displayed as comparable as possible to
visualize how the introduction of dimethyl groups altered the orientation of the pendant
arms, (Fig 43).
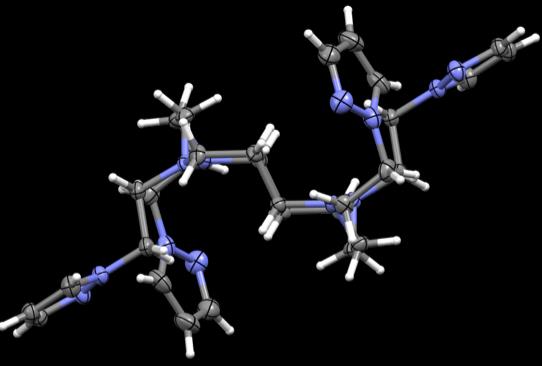
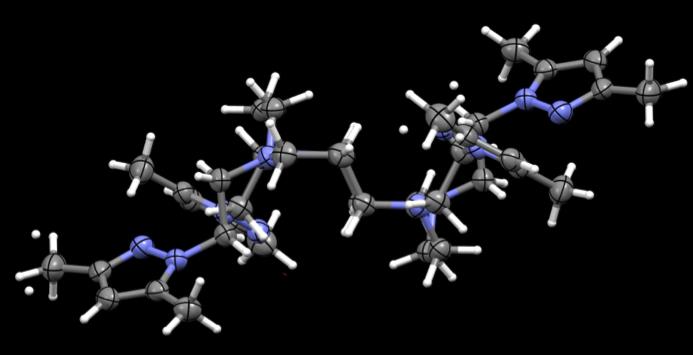
Figure 43. Crystal structures of PzCm and DiMePzCm for comparison.
It can be seen how the addition of dimethylated pyrazole arms alters the orientation of
the ligand in general in Fig 43, at least in the solid state. PzCm shows pendant arms 1
and 3 to be expanded within the same general plane as the macrocycle where arms 2 and
4 seem to flex up and out of the plane. In the dimethylated analogue, arms 1 and 3 still
seem to be in the same field but slightly twisted at an angle where arms 2 and 4 are still
flexed out of field, but also twisted at an angle. From this comparison, it appears that
when the methylated pendant groups are added, the pendant arms seem to twist at an
angle compared to the unmethylated analogue.
A comparison of the PzCn and DiMePzCn crystal structures was also examined,
(Fig 44). In PzCn, arms 1 and 4 are in the plane of the ligand but at an angle where arms
2 and 3 are flexed out of plane at an angle that appears perpendicular to the ligand.
DiMePzCn arms 1 and 4 are extended in the same plane as the ligand and arms 2 and 3
are outstretched out of the plane of the macrocycle. In the instance of DiMePzCm, it
seemed the dimethyl groups increased the angle where in the case of DiMePzCn, the
dimethyl groups decreased the angle of the arms to the ligand.
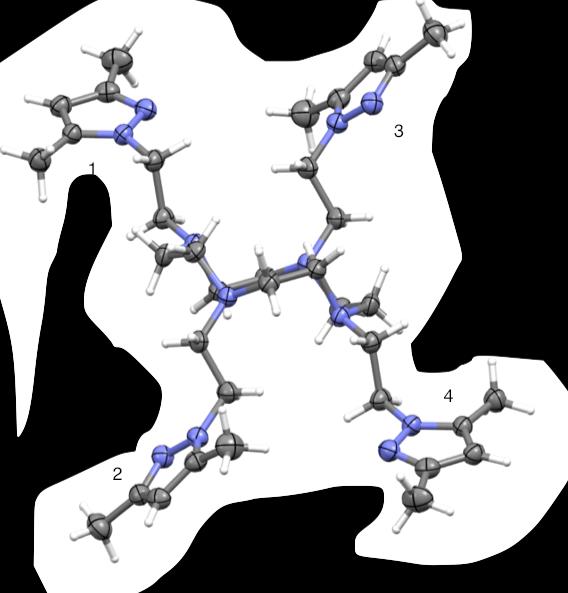
Figure 44. Crystal structures of PzCn and DiMePzCn for comparison.
3.7 PĪ Space Group
PĪ defines the space group symmetry of all four crystal structures. P1 itself describes
no symmetry; however, PĪ accounts for inversion symmetry. Inversion symmetry occurs
when there is one point of inversion within the unit cell where the opposite sides are
symmetrical to the other.54 When solving the X-ray structure, if PĪ symmetry is
represented, then using the inversion point one half of the unit cell can be created from
the other. In the case of all of the ligand structures, an inversion center lies in the center
of the structure, since there is only one molecule per unit cell.
3.8 Synthetic Considerations
Of the brominated and chlorinated versions of the pyrazole pendant arm precursors, it
was determined the brominated ethylpyrazoles were preferred. The chlorinated versions
needed to undergo distillation to purify the product, where the brominated product did not
have to undergo this process. It also took two days for the pendant arms to be added with
the bromide leaving group, where it took seven days for the chlorinated pendant arms to
react. Though the brominated reactions did need another aliquot of BP or DiMeBP to be
added after 24 hours due to the decomposition before the reaction was completed, this
was the only factor that played into the two-day length of the reaction. As a result,
bromoethylpyrazole/3,5-dimethylpyrazole was used when possible rather than the
chlorinated analogues.
DiMePzCm, PzCm, and PzCn were all directly crystallized from the worked-up
reaction mixture. DiMePzCn provided the most difficult to purify. The crystals were
eventually purified after multiple chromatography trials, TLC, and multiple NMR's. The
solid from this reaction was insoluble in most solutions and refused to crystalize out due
to the large levels of impurities in the crude product. In order to rid the final product of
any lingering impurity, a column was performed. Silica columns were attempted at first,
but seemed to maintain some of the product and usually were deemed unsuccessful. Due
to these prior attempts, it was decided to use alumina in place of typical silica gel.
The alumina column was performed using methylene chloride and 2-15% methanol.
A total of 60 fractions were collected and each fraction was individually TLC'd. From
these TLC plates, it appeared that fractions 8-16 held product where the other fractions
held impurity. These "pure" fractions were collected, dried, and worked up using
methylene chloride and basic water (ammonia hydroxide).
3.9 Metal Ion Complex Attempts
Attempts were made to synthesize metal ion complexes from the previously
synthesized pyrazole pendant macrocyclic ligands. Unfortunately, no attempt proved
successful as each reaction yielded no crystalline product and ultimately no useful spectra
to suggest even a small amount of the reaction had worked as hoped. To try and pinpoint
what was and was not working, several reactions were performed using a different
macrocycles, different solvents, varying amounts of said solvents, and reaction mixtures
left for extended crystallization times (Table 14).
After several failed reactions, mass spectrometry was performed to decide whether
the reactions were working at some level. Due to the immediate color change noted when
ligand was added to metals, it was assumed a reaction was taking place, but it was not
immediately clear as to what the product was. However, this mass spec showed only the
ligand, with either one or two pyrazole arms dissociated.
Combined, these results led to the belief that maybe molecular complexes were not
formed, but possibly polymeric complexes. If that were the case, then the ligand would
be coordinating to multiple metal ions, which might have been ligated to multiple
ligands. A strong consideration is that the previously synthesized ligands may have too
many coordinating arms to properly ligate to a metal ion and to form a molecular
complex. Since transition metal ions are acidic in nature, it should also be considered that
when attempting to coordinate to a metal, the metal ion is actually aiding in the
dissociation of a pendant arm(s) from the macrocycle.
There are previously reported findings that use macrocycles coordinated with three
pendant arms that are successful when complexing with a metal ion.7 This supports the
possibility that four coordinating arms may be too many.
Table 14. Attempted metal ion reactions
Metal complex
Color change
DiMePzCm
2-methoxyethanol
dimethylformamide
Iron (II) tetrafluoroborate
Copper perchlorate
DiMePzCm
Chapter 4. Conclusions and Future Directions
4.0 Conclusions
After referring to the Attarian et al paper for synthesis of 2-(1-chloroethyl)pyrazoles
(and ultimately 2-(1-bromoethyl)pyrazoles), it was concluded that either set of
compounds worked well for the synthesis of the pyrazole pendant tetraazamacrocycles.53
Though both the brominated and the chlorinated compounds could be used for synthesis,
the brominated precursors were preferred. This was primarily due to the more time-
efficient synthesis; two days versus seven days for the chlorinated compounds.
The four structures, PzCm, PzCn, DiMePzCm, and DiMePzCn were successfully
synthesized and characterized. However, attempts to form complexes with metal ion were
deemed unsuccessful. When mass spectrometry was performed on one complex reaction
mixture containing cobalt (II) chloride hexahydrate, there were peaks indicative of a
pendant arm falling off. This suggests that the ligands might be sensitive to acid, so
caution is necessary when forming metal ion complexes.
For the complexes, different metal ions in different forms were obtained for synthesis.
Unfortunately, no crystals were formed over a two month period and nothing to suggest a
molecular complex was identified. A variety of solvents were also used to attempt
complex formation. Again, varying solvents with varied metal ions did not yield any
productive results.
For future directions, since complexes of macrocyclic ligands do hold promise in
medicinal and biomedical applications, there are a large number of other metals and
solvents that can be used to attempt complex synthesis. One factor that may be causing
the complex to not form properly could be that there are four pendant arms, which may
be too many, so a ligand with 2 or three coordinating pendant arms may be more suitable.
1. Lindoy, L. F. The Chemistry Of Macrocyclic Ligand Complexes; Cambridge
University Press: 1989, pp 1–15.
2. Mewis, R. E.; Archibald, S. J. Biomedical applications of macrocyclic ligand
complexes. Coord. Chem. Rev. 2010, 254, 1686–1712.
3. Busch, D. H. Distinctive coordination chemistry and biological significance of
complexes with macrocyclic ligands. Acc. Chem. Res. 1978, 11, 392-400.
4. Delgado, R.; Felix, V.; Lima, L. M. P.; Price, D. W. Metal complexes of cyclen and
cyclam derivatives useful for medical applications: a discussion based on
thermodynamic stability constants and structural data. Dalton Trans. 2007, 2734-
5. Liang, X.; Sadler, P. J. Cylam Complexs and Their Applications in Medicine. Chem.
Soc. Rev. 2004, 33, 246-266.
6. Nirmala, G.; Kalilur Rahiman, A.; Sreedaran, S.; Jegadeesh, R.; Raaman, N.;
Narayanan, V. N-Benzoylated 1,4,8,11-Tetraazacyclotetradecane and Their
Copper(II) Andnickel(II) Complexes: Spectral, Magnetic, Electrochemical, Crystal
Structure,Catalytic and Antimicrobial Studies. Spectrochim. Acta. A. Mol. Biomol.
Spectrosc. 2010, 77,92–100.
7. Dorazio, S. J.; Olatunde, A. O.; Tsitovich, P. B.; Morrow, J. R. Comparison Of
Divalent Transition Metal Ion ParaCEST MRI Contrast Agents. J. Biol. Inorg. Chem.
2014, 19,191–205.
8. Habibi, D.; Nabavi, J.; Heydari, S.; Vakili, S. Synthesis Of a New Cyclen-Based
Compound as a Potent Anti-Tumor Medicine. Orient. J. Chem. 2013.
9. Jiang, Z. X.; Yu, B. Y. The Design and Synthesis of Highly Branched and Spherically
Symmetric Fluorinated Macrocyclic Chelators. Synthesis (Stuttg). 2008, 2, 215-220.
10. Norouzy, A.; Nau, W. Synthetic macrocyclic receptors as tools in drug delivery and
drug discovery. Drug Target Rev. 2014.
11. Yudin, A. K. Macrocycles: lessons from the distant past, recent developments, and
future directions. Chem. Sci. 2015, 6, 30-49
12. Mallinson, J.; Collins, I. Macrocycles in new drug discovery. Future Med. Chem.
2012, 4, 1409-1438.
13. Zhang, C. X.; Lippard, S. J. New metal complexes as potential therapeutics. Curr.
Opin. Chem. Biol. 2003, 7, 481-489.
14. Bush AI: Metal complexing agents as therapies for Alzheimer's disease. Neurobiol.
Aging. 2002, 23, 1031-1038.
15. Heffern, M. C.; Yamamoto, N.; Holbrook, R.J.; Eckermann, A. L.; Meade, T. J.
Cobalt Derivatives as Promising Therapeutic Agents. Curr. Opin. Chem. Biol. 2013,
17, 189-196.
16. Hubin, T. I. J.; McCormick, J. M.; Collinson, S. R.; Alcock, N. W.; Busch, D. H.
Ultra Rigid Cross-Bridged Tetraazamacrocycles as Ligands—the Challenge And. J.
Chem. Soc. Chem. Commun. 1998: 1675-1676.
17. Milgrom, L. R. The Colours Of Life: An Introduction to the Chemistry of Porphyrins
and Related Compounds. Oxford University Press: 1997.
18. Hubin, T. J.; McCormick J. M.; Collinson S. R.; Alcock N.W.; Busch D. H.;
Synthesis and coordination chemistry of topologically constrained azamacrocycles. J.
Chem. Soc. Chem. Commun. 2002, 241, 27.
19. G. Jori and E. Reddi, The role of lipoproteins in the delivery of tumour-targeting
photosensitizers. Int. J. Biochem. 1993, 25, 1369-1375.
20. Larson, Delmar. 5.10: Chelate and macrocyclic effects.
(accessed May 11, 2016).
21. Cram D. J.; Degrandpre M.; Knobler C. B.; Trueblood K. N. J. Am. Chem. Soc.,
1984, 106, 3286.
22. Wadas, T. J.; Wong, E. H.; Weisman, G. R.; Anderson, C. J. Copper chelation
chemistry and its role in copper radiopharmaceuticals. Curr. Pharm. Des. 2007, 13, 3-
23. Pechmann, H. V. Pyrazol aus Acetylen und Diazomethan. Chem. Ber. 1898, 31,
24. Malinowska, K.; Galecka, E.; Modranka, R.; Rutkowski, M.; Kedziora, J. Derivatives
pyrazoles used in a medical treatment. Pol. Merku.r Lekarski. 2008, 24, 151-157.
25. Naim, M. J.; Alam, O.; Nawaz, F.; Alam, J.; Alam, P. Current status of pyrazole and
its biological activities. J. Pharm. Bioallied. Sci. 2016, 8, 2-17.
26. Costamagna, J.; Ferraudi, G., Matsuhiro, B. Complexes of macrocycles with pendant
arms as models for biological molecules. Coord. Chem. Rev. 2000, 196, 125–164.
27. University of Waterloo. Coordination Numbers.
28. Wikipedia. Cubic crystal sy
(accessed May 11, 2016).
(accessed May 5, 2016).
30. Lauterbur, P. C. Image Formation by Induced Local Interactions: Examples of
Employing Nuclear Magnetic Resonance. Nature. 1973, 242, 190–1.
31. Edelman, R. R.; Warach, S. Magnetic Resonance Imaging. N. Engl. J. Med. 1993,
328, 708-716.
32. Berger, A. Magnetic resonance imaging. BMJ. 2002, 324, 35.
33. Siemens Healthcare. Magnetom Essenza.
(accessed May 5, 2016).
34. Brown, M. A.; Semelka, R. C. MRI: Basic Principles and Applications; Wiley-Liss,
3rd edition: 2003.
35. Rinck P. Magnetic Resonance in Medicine. The Basic Textbook of the European
Magnetic Resonance Forum; The Round Table Foundation, 9th edition: 2016.
36. MRI Master. Histroy(accessed May 10,
37. Karabulut, N. Gadolinium deposition in the brain: another concern regarding
gadolinium-based contrast agents. Diagn. Interv. Radiol. 2015, 21, 269-270.
38. Zhou, Z.; Lu, Z. R. Gadolinium-Based Contrast Agents for MR Cancer Imaging.
Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 1-18.
39. Olatunde, A.; Cox, J.M.,; Daddario, M.D.; Spernyak, J.A.; Benedict, J.B.; Morrow,
J.R. Seven-Coordinate CoII, FeII and Six-Coordinate NiII Amide-Appended
Macrocyclic Complexes as ParaCEST Agents in Biological Media. J. Inorg. Chem.
2014, 53, 8311-8321.
40. Boundless. Diamagnetism and Paramagnetism.
(accessed May 5, 2016).
41. Wikipedia. Magnetic Resonance Imaging.
(accessed May 5, 2016).
42. Elster, A. D. Paramagnetic Relaxation-how does gadolinium cause relaxtion.
Questions and Answers in MRI
(accessed May 11, 2016).
43. Miessler G. L.; Tarr, D. A. Inorganic Chemistry; Pearson/Prentice Hall, 3rd Ed: 2003.
44. Woods, M.; Donald, E. W. C.; Sherry, A. D. Paramagnetic lanthanide complexes as
PARACEST agents for medical imaging. Chem. Soc. Rev. 2006, 35, 500-511
45. Wu, Y.; Zhou, Y.; Quari, O.; Woods, M.; Zhao, P.; Soesbe, T. C.; Kiefer, G. E.;
Sherry, A. D. Polymeric PARACEST Agents for Enhancing MRI Contrast
Sensitivity. J. Am. Chem. Soc. 2008, 130, 13854-13855.
46. Dorazio, S. J.; Tsitovich, P. B.; Siters, K. E.; Spernyak, J. A.; Morrow, J. R. Iron(II)
PARACEST MRI Contrast Agents. J. Am. Chem. Soc. 2011, 133, 14154-14156.
47. Chong, H. S.; Garmestani, K.; Bryant, L. H.; Milenic, D. E.; Overstreet, T.; Birch, N.;
Le, T.; Brady, E. D.; Brechbiel, M. W. Synthesis and evaluation of novel macrocyclic
and acyclic ligands as contrast enhancement agents for magnetic resonance imaging.
J. Med. Chem. 2006, 49, 2055-2062.
48. Seeger, A.; Kramer, U.; Fenchel, M.; Grimm, F.; Bretschneider, C.; Doring, J.;
Klumpp, B.; Tepe, G.; Rittig, K.; Seidensticker, P. R.; Claussen, C. D.; Miller, S.
Comparison between a linear versus a macrocyclic contrast agent for whole body MR
angiography in a clinical routine setting. J. Cardiovas. Mag. Res. 2008, 10, 63.
49. Sheldrick, G. M. SADABS, version 2007/4; University of Göttingen: Göttingen,
50. Altomare, A.; Burla, M. C.; Camalli, M.; Cascarano, G. L.; Giacovazzo, C.;
Guagliardi, A.; Moliterni, A. G. G.; Polidori, G.; Spagna, R. SIR97: A new
program for solving and refining crystal structures; Istituto di Cristallografia, CNR:
Bari, Italy, 1999.
51. Sheldrick, G. M. Acta. Cryst. 2008, A64, 112. DOI:10.1107/S0108767307043930
52. Lopez, P.; Seipelt, C. G.; Merkling, P.; Sturz, L.; Alvarez, J.; Dolle, A.; Zeidler,
M.D.; Cerdan, S.; Ballesteros, P. N-2-(Azol-1(2)-yl)ethyliminodiacetic Acids: A
Novel Series of Gd(III) Chelators as T2 Relaxation Agents for Magnetic Resonance
Imaging. Biorg. Med. Chem. 1998, 7, 517-527.
53. Attarian, O.S; et al. Synthesis of N-Vinylpyrazoles. Chem. Heterocyclic Comp. 2004,
41, 452-455.
54. Cockcroft, J. K.; Driessen, H. Triclinic Space Groups. Birkbeck College, University
of L(accessed May 5, 2016).
Appendix A – 2D NMR of PzCm
Appendix B – 2D NMR of PzCn
Appendix C – 2D NMR of DiMePzCm
Appendix D – 2D NMR of DiMePzCn
Source: http://digitalcommons.buffalostate.edu/cgi/viewcontent.cgi?article=1010&context=forensic_science_theses
Esta es una publicación del Instituto de Cardiología de Corrientes "Juana F. Cabral " y la FUNDACION CARDIOLOGICA CORRENTINA PUBLICACION Vuelve la Clínica Intensiva para Bajar de Peso GRATUITA Año 7 Nº54 Recientemente se ha llevado a cabo en la ciudad de Buenos Aires el JUNIO 2008 Congreso Mundial de Cardiología, que reunió a 15000 especialistas de todo el mundo y del que muchos miembros del Instituto de Cardio-logía hemos tenido el honor de participar.Sin lugar a duda, una de las afirmaciones mas contundentes de este congreso ha sido justamente el título de esta nota: "La obesidad es el factor de riesgo más importante para el corazón".
IASGO HPB AND GI ONCOLOGY POST-GRADUATE COURSE 56 STRATEGIES FOR DIFFERENTIATING XANTHOGRANULOMATOUS CHOLECYSTITIS FROM GALL BLADDER CARCINOMA- A TERTIARY CARE CENTRE EXPERIENCE Authors: Kishore Rajaguru1, Samiran Nundy2 1Jurong Health, 2Sir Gangaram Hospital Abstract: Aim Xanthogranulomatous cholecystitis (XGC) mimics Gallbladder carcinoma (GBC) in both preoperative and intra-operative settings and the patient may undergo an unnecessary radical cholecystectomy which is associated with a greater morbidityrather than a cholecystectomy alone.We postulated that a pre-operative diagnosis of XGC might benefit patients by avoiding radical procedures and attempted to identify the features of XGC which would differentiate it from GBC before and during surgery. Methods All the patients who underwent gall bladder related operations (benign and malignant),over a period of 5 years from 2010 to 2014 were reviewed in a tertiary centre hospital in Delhi, India.After the histopathological reports the patients were placed into two groups - those with XGC and GBC.The following parameters were recorded–clinical features,biochemical and radiological findings including the presence of gall stones, common bile duct (CBD) stones,focal or diffuse wall thickening of the GB,the presence of intramural bands or nodules in the wall, lymph node enlargement,mucosal enhancement,and the status of the interface between the liver bed and gall bladder. A comparison was made between the groups. Results Patients with a long history of recurrent abdominal pain and who on imaging were found to have a diffusely thickened gall bladder wall, with cholelithiasis, choledocholithiasis and submucosal hypoattenuated nodules were likely to have XGC while those with anorexia,weight loss,focal thickening of the gallbladder wall on imaging and dense local organ infiltration were more likely to have GBC. The presence of lymph nodes on imaging and the loss of a fat plane interface between the liver and gallbladder were not differentiating factors. Conclusion The differentiation of XGC and GBC pre-operatively remains a challenge but is possible via certain clinical and imaging characteristics.However the definitive diagnosis still remains a frozen section histopathological examination to avoid a radical resection in patients who have a benign condition.

