Psh.org.pk

PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
Volume: 1 0, No.1 Jan – Mar 2016
President's Column
Dear Colleagues Asalam-o-Alikum ! As the president of PSH it gives me great pleasure to write few words. One of the major reasons for establishing the Pakistan Society of Haematology was to endorse due recognition to the holding yearly conferences and by teaching practical applications in Haematology in workshops and symposium. After decades of work, the last two to three years have brought about a successful translation of basic immunology that gave rise to two promising strategies to modify the immune system
Major General
to treat cancer. One approach involves antibodies treat
Mohammad Ayyub, HI(M)
cancer. One approach involves antibodies targeting immune
checkpoints, CTLA-4 and PD-1, to release the brake on T cells to attack tumor cells. Another strategy involves infusion of T cells genetically modified to express a chimeric antigen receptor (CAR) to target a specific tumor antigen. While immune checkpoint inhibitors gained their first clinical success in solid tumors, CAR T cells first emerged as a promising therapy in hematologic malignancies after several clinical trials demonstrating unprecedented clinical activity of CD19-targeted CAR T cells in patients with relapsed/refractory acute lymphocytic leukemia (ALL). CD19-targeted CAR T cells have emerged as one of the most potent therapeutic agents in ALL and serve as a strong proof of principle for this novel immune-based approach. Multicenter phase II trials are commencing shortly in both pediatric and adult ALL. Furthermore, many groups are exploring the feasibility of utilizing CAR T cells to target non-C19 positive malignancies such as acute myeloid leukemia, multiple myeloma, brain tumors, prostate cancer, mesothelioma, and ovarian cancer, and more reports from these studies are expected in 2016. These new strategies of treatment in the field of Haematology are eye opener to our young budding haematologists, who are the future hope of Pakistan.
With warm regards,
Major General Mohammad Ayyub, HI(M)
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
ACADEMICS
PRIMARY MULTIPLE PLASMACYTOMAS
Raheel Iftikhar, Parvez Ahmed, Tariq Mahmood Satti, Qamar un Nisa Chaudhry, Syed Kamran Ahmed,
Mehreen Ali Khan, Armed Forces Bone Marrow Transplant Centre, Rawalpindi – Pakistan
Introduction
Plasma cell neoplasms are a group of disorders characterized by the abnormal
proliferation of a single clone of plasma cells, typically producing a monoclonal
immunoglobulin. Plasma cell neoplasms can present as a solitary
plasmacytoma or multiple myeloma Solitary plasmacytomas most frequently
occur in bone (plasmacytoma of bone), but can also be found outside bone in
soft tissues (extramedullary plasmacytoma). Multiple extramedullary
plasmacytomas are very uncommon with only few documented case reports
in literature.
Case Report
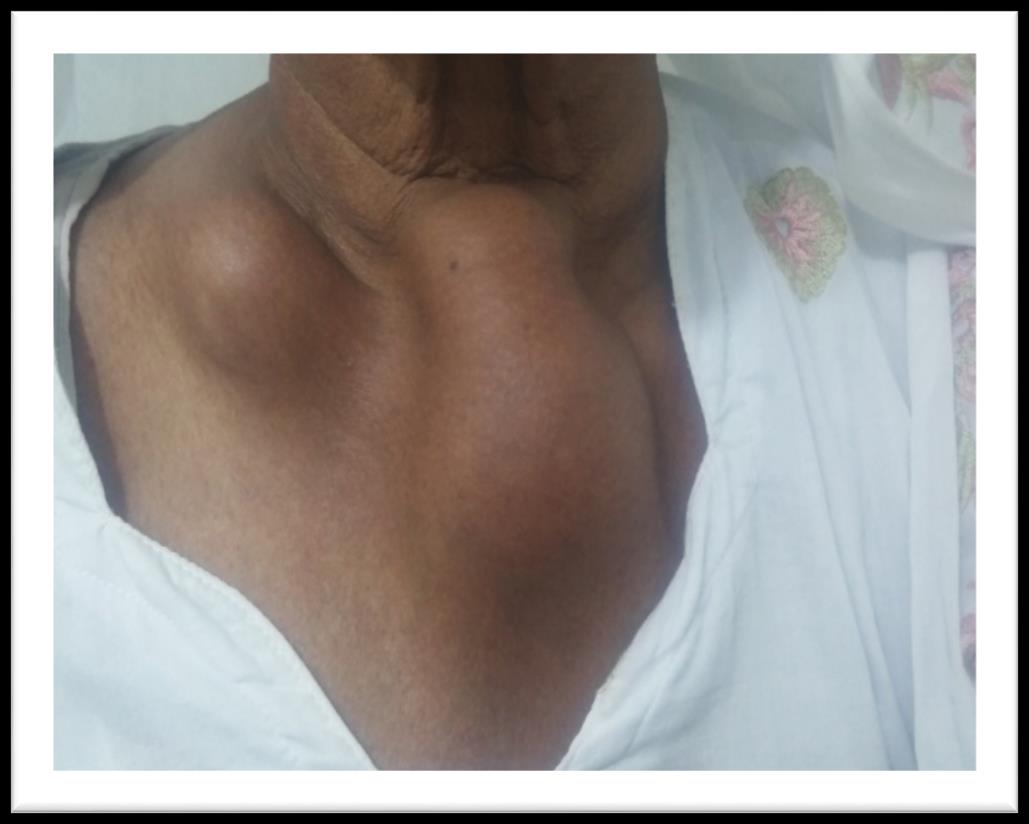
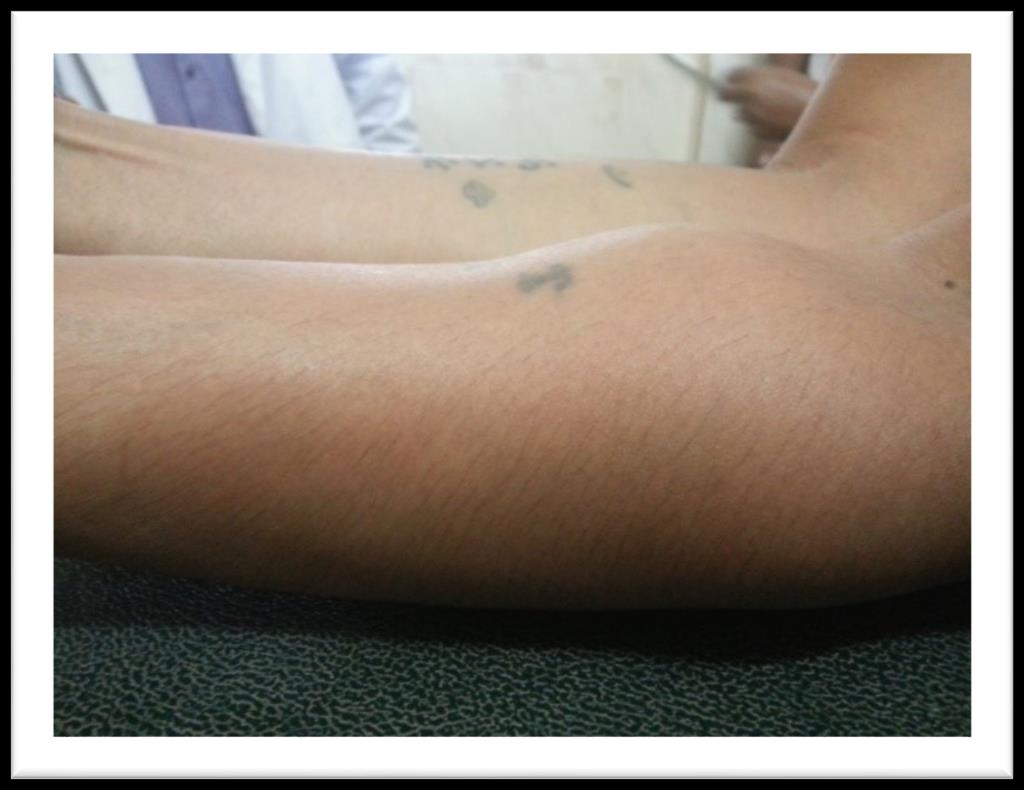
A 60 years old house wife presented in outpatient department with history of gradually increasing swellings over chest, left forearm and right thigh for last 6 months. She had first noticed this swelling over front of her chest. It was painless and around size of a lemon. It gradually increased over next 3 months and patient noticed appearance of similar swellings over left forearm and right thigh. She also developed easy fatigability and inability to carryout routine physical activity. Past history was significant for hypertension for last 6 years for which she was taking tablet losartan off and on. Rest of her personal, family, socioeconomic and drug history was unremarkable. On examination she had a blood pressure of 140/90 mmHg. She had visible swellings over middle part of right clavicle, upper sternum, right thigh and left forearm (Figure 1). Swellings were fixed to underlying structures, firm to hard in consistency, had a smooth surface, non-pulsatile, non-tender without overlying skin erythema and were not fluctuant. Swelling over clavicle was 3 x4 cm, sternal swelling was 8 x 4 cm, forearm swelling was 3 x 2 cm (Figure 2) while thigh swelling was 4 x 4 cm. Rest of her general and systemic examination was unremarkable. Her complete blood counts showed hemoglobin, WBC and platelets within normal range. Erythrocyte sedimentation rate was 120 mm fall
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
at 1 hour with rouleaux formation visible on peripheral smear. Her liver
functions tests, renal function tests, Urine microscopy, Serum lactate
dehydrogenase, serum Albumin levels were all with in normal range. X-ray of
Chest, forearm and thigh were unremarkable except for soft tissue masses
visible in affected area on radiographs in chest, right femur and left forearm.
CT scan of chest showed lytic lesions in right clavicle and upper sternum
extending into overlying skin and soft tissue. CT scan of left forearm and right
thigh demonstrated soft tissue non enhancing lesions with underlying bone
involvement. Fine needle aspiration of Sternal mass and forearm swelling was
done both of which showed sheets of mature plasma cells with abundant
basophilic cytoplasm. The nucleus was round and eccentrically located with
marked perinuclear hof consistent with diagnosis of plasmacytoma. These
cells were CD138 positive and lambda light chain restricted. Her bone marrow
aspiration and trephine biopsy showed only 3 % plasma cells. Serum protein
electrophoresis showed presence of sharp monoclonal band in gamma region
which on immunofixation was IgG lambda. On quantification M-protein was
1.48 g/dl. Serum B2 microglobulins, Serum free light chains and skeletal
survey was unremarkable except for above defined lesions. On the basis of
above history and investigations patient was diagnosed as having multiple
solitary plasmacytomas with minimal marrow involvement as per definition of
International Myeloma Working Group 2014 updated criteria for the diagnosis
of multiple myeloma (3). Because of multiple plasmacytomas at different sites
and high risk of progression to Multiple myeloma, Patient was advised
combination chemotherapy with melphalan, thalidomide and prednisolone.
Discussion
Solitary plasmacytoma constitute approximately 5 percent of all cases of plasma cell disorders. Plasmacytoma is an abnormal proliferation of plasma cells and can be primary or secondary (4). Primary plasmacytomas occur in absence of pre-existing pathology while secondary plasmacytomas can appear after treatment of multiple myeloma or after radiotherapy of Solitary plasmacytoma. Men are diagnosed twice as frequently as women. The median age at diagnosis is 55 to 65 years (5). These plasmacytomas can be osseous (medullary) or nonosseous (extramedullary) tissues. Extramedullary plasmacytomas arises from outside bone marrow and are usually solitary,
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
however, rarely multiple plasmacytomas are present. Sites most commonly
involved in extramedullary plasmacytoma are aerodigestive tract,
gastrointestinal tract, urogenital tract, skin, soft tissues, lung and breast. The
International Myeloma Working Group in 2003 recognized a separate
classification of plasmacytomas that occur as multiple sites of disease in soft
tissue, bone, or both soft tissue and bone as multiple solitary
plasmacytoma. There diagnostic criteria includes : No or small amount of M-
protein in serum and/or urine, more than one localized area of bone
destruction or extramedullary tumour of clonal plasma cells, otherwise normal
skeletal survey and no myeloma related organ damage (6). These patients can
have isolated plasmacytomas without any monoclonal gammopathy or can
have small amount of detectable paraprotein. Patients with multiple
plasmacytomas have aggressive disease with high risk of progression to
multiple myeloma. In patients with two concurrent discrete plasmacytomas
with no bone marrow involvement radiation to both sites is recommended. In
patients with more than two concurrent lesions, systemic therapy identical to
that used for multiple myeloma is indicated, even if the bone marrow is normal
(7). For patients who develop two or three solitary lesions within a period of
one to two years, subsequent therapy should be as if the patient has multiple
myeloma.
Hussain A and colleagues reported a case of multiple extramedullary
plasmacytomas with lytic bone lesions (8). Tüting and Bork published a case
of a solitary cutaneous plasmacytoma of left thigh without bone marrow
involvement (9). Kaviani et al. reported case of a 70-year-old woman with
bilateral breast masses that afterwards proved to be a recurrence from
extramedullary plasmacytoma of the mediastinum for which she got treatment
5 years back (10) . Shahid et al. presented a rare case of solitary lytic lesion
in the medial part of clavicle with biopsy revealing plasmacytoma (11). Most
patients with SPB progress to multiple myeloma in 4 years. In contrast, 80 %
Patients with multiple plasmacytoma progress to multiple myelmoa in 1 year
(12). It can be concluded that plasma cell disorders can rarely present as
multiple extramedullary plasmacytoma, these patients have more aggressive
disease course and needs to be treated like symptomatic multiple myeloma.
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
Figure 1: Plasmacytomas of right clavicle and sternum.
Figure 2: Plasmacytomas of left forearm.
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
References
Hughes M, Doig A, Soutar R. Solitary plasmacytoma and multiple myeloma: adhesion molecule and chemokine receptor expression patterns. Br J Haematol 2007; 137:486.
Reed V, Shah J, Medeiros LJ, Ha CS, Mazloom A, Weber DM, Arzu IY, Orlowski RZ, Thomas SK, Shihadeh F, Alexanian R. Solitary plasmacytomas. Cancer. 2011 Oct 1;117(19):4468-74.
Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, Kumar S, Hillengass J, Kastritis E, Richardson P, Landgren O. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. The Lancet Oncology. 2014 Nov 30;15(12):e538-48.
Soutar R, Lucraft H, Jackson G, et al. Guidelines on the diagnosis and management of solitary plasmacytoma of bone and solitary extramedullary plasmacytoma. Br J Haematol 2004; 124:717.
Warsame R, Gertz MA, Lacy MQ. Trends and outcomes of modern staging of solitary plasmacytoma of bone. Am J Hematol 2012; 87:647.
International Myeloma Working Group, "Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group,"British Journal of Haematology, vol. 121, pp. 749–757, 2003.
Kilciksiz S, Karakoyun-Celik O, Agaoglu FY, Haydaroglu A. A review for solitary plasmacytoma of bone and extramedullary plasmacytoma. The Scientific World Journal. 2012;2012.
Hussain A, Singh M, Singh K, Bagga H. Multiple Extramedullary Plasmacytoma with Lytic Bony Lesions: A Rare Case Report. Case reports in medicine. 2013;2013.
T. Tüting and K. Bork, "Primary plasmacytoma of the skin," Journal of the American Academy of Dermatology, vol. 34, no. 2, part 2, pp. 386–390, 1996
10. A. Kaviani, M. D. Zavareie, M. Noparast, and S. Keyhani-Rofagha, "Recurrence of
primary extramedullary plasmacytoma in breast both simulating primary breast carcinoma," World Journal of Surgical Oncology, vol. 2, article 29, 2004
11. M. Shahid, N. Z. Hali, A. Mubeen, et al., "Solitary plasmacytoma of clavicle—a rare
case presentation,"Journal of Cytology & Histology, vol. 1, article 103, 2010.
12. Hill QA, Rawstron AC, de Tute RM, Owen RG. Outcome prediction in plasmacytoma
of bone: a risk model utilizing bone marrow flow cytometry and light-chain analysis. Blood 2014; 124:1296.
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
CRYSTALLINE BONE MARROW IN YOUNG LADY
Farwa Raza, Ch Altaf Hussain, Saleem Ahmed Khan, Hamid Saeed, Mohammad Ayyub Department of
Pathology, Armed Forces Institute of Pathology, Rawalpindi
Introduction:
Hyperoxaluria is found in approximately 20% of stone formers. Dietry factors
such as high oxalate and low calcium intake account for hyperoxaluria in most
such patients. The degree of hyperoxaluria observed in these circumstances
is mild. Primary hyperoxaluria is a rare autosomal recessive disorder of hepatic
glyoxylate metabolism resulting in overproduction of oxalate. Oxalate
nephropathy results in renal failure, which leads to further increase in plasma
concentration of oxalate. Calcium oxalate can then deposit into wide array of
tissue including bone, retina, peripheral nerves, arterial media and the heart.
In patients with primary hyperoxaluria, kidney failure can occur at any age from
infancy to middle or even late adulthood. Earlier literature showed that CKD
develops in 50% of patients by 15 years of age and 80% develop renal failure
by age of 30 years. Recent studies suggest that with improved diagnosis and
management, median age at kidney failure is 33 years. Due to lack of
familiarity with disease, delay of many years from onset of symptoms to
diagnosis is common. Onset of calcium oxalate stone formation or
nephrocalcinosis in childhood and adolescence that are associated with CKD
are important clues in diagnosis and warrant specific diagnostic testing for the
disease. Here we report a case of oxalosis who presented with end stage renal
failure and erythropoietin resistant anaemia, with oxalate crystal deposition in
bone marrow.
Case Report:
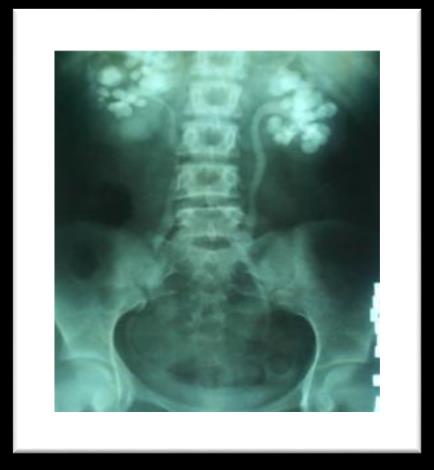
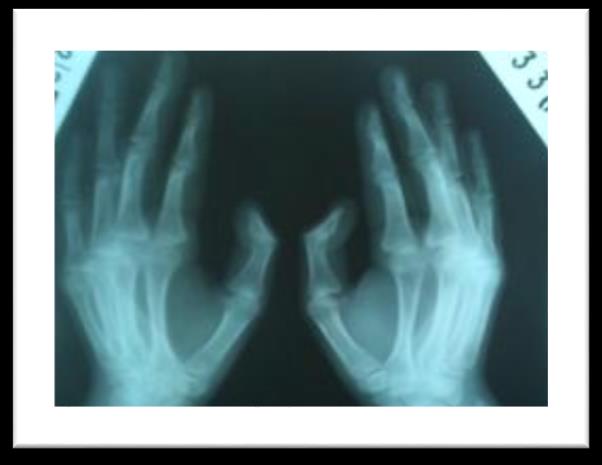
A 25 year old female patient diagnosed case of CKD on regular haemodialysis since 2012. She was referred to our department for evaluation of refractory anaemia. She had past history of multiple stones in both kidneys (figure 1) & was operated in emergency in 2003. Multiple stones were removed from left kidney, the maximum size of stone was 2-2.5cm. Patient was investigated later on and found to be hyperparathyroid and stone chemical analysis revealed that they were calcium oxalate. Radiological evaluation showed patient had osteopenia (figure 2).
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
Figure 1: Bilateral multiple stones
Figure 2: Osteopenia
She was lost to follow up till 2012 when she again reported to urology OPD & was diagnosed as CKD & started with regular haemodialysis twice weekly, taking Vitamin D, Calcium supplementation, antihypertensive treatment and erythropoietin injection with regular monitoring of RFTs, Bone profile & Parathyroid hormone. Patient was referred to Haematology department for evaluation of erythropoietin resistant anaemia. She had four siblings and all are alive & healthy. Multiple blood transfusions had been given since 2012 & last transfusion was 2 weeks back. On examination she was thin lean, pale looking young lady, there was no visceromegaly. Her full blood counts showed Hb: 8.4 gm/dl, WBC count: 1.92 x 109 /L, Plt count: 319 x 109 /L. Her serum urea: 8.4 mmol/l (N 3.3-6.7 mmol/l); serum creatinine: 304 umol/l (N 55-100umol/l); Serum albumin: 41g/l (N 35-50g/l) and s.alkaline phosphatase: 788U/L (N <258U/l). Bone profile showed serum calcium was 2.26 mmol/l (N 2.1-2.65 mmol/l); serum phosphate: 3.0 mmol/l (N 0.81-1.62mmol/l) and total Vitamin D: 23nmol/l (deficiency <25 nmol/l). USG & CT Scan abdomen showed both kidneys were atrophic with with multiple calculi in right kidney. Peripheral blood film & bone marrow aspirate showed bicytopenia. Histological examination of
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016

PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
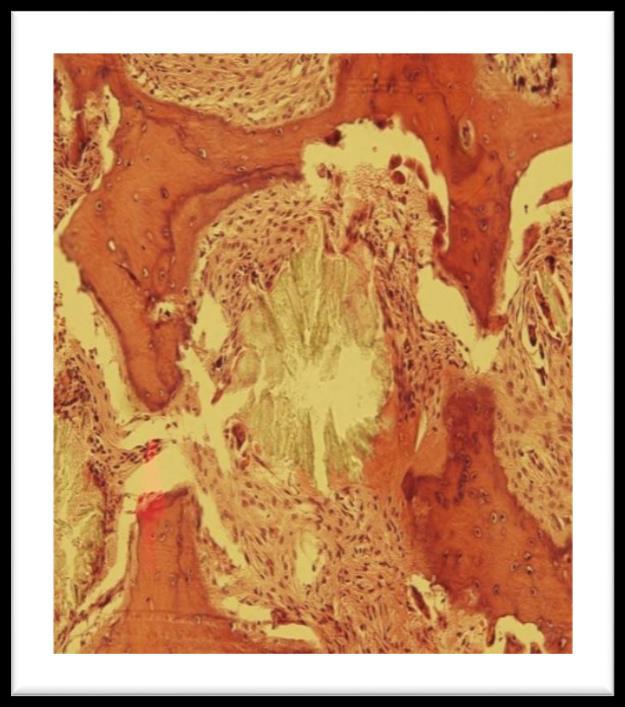
trephine marrow biopsy showed pericrystalline giant cell granulomata with foreign body giant cells and moderate paratrabecular fibrosis. There is heavy deposition of oxalate crystals arranged in radial pattern around central amorphous core. These crystals were birefringent under polarized light. Patient was further investigated for gene mutation.
Figure 1: Bone marrow trephine showing pericrystalline giant cell granulomata
Figure 2: Oxalate crystals seen through polarized light
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
Discussion:
In the patient with hyperoxaluria, symptoms may occur any time from birth to adulthood and presentation can vary from mild to severe. The first sign is usually blood in urine, pain, passage of stone or urinary tract infection related to kidney stones. Minority of patient presents with kidney failure as first sign. Our patient was young adult who presented rather late with wide spread deposition of oxalates, as evidenced by nephrolithiasis, renal failure and morphological evidence of oxalate deposition in bone and bone marrow. Even though family history of patient was not significant, patient was labeled to have late onset Primary Hyperoxaluria. Anaemia in this patient has been attributed to chronic renal failure and bone marrow replacement by pericrystalline granulomata. Hyperoxaluria may be primary, secondary or idiopathic. Secondary hyperoxaluria results from impaired renal excretion, excessive dietry intake of oxalate with ascorbic acid and ethylene glycol ingestion and increased absorption in patients with chronic inflammatory bowel disease. Primary hyperoxalurias (PH1 and PH2) are autosomal recessive disorders that result from inherited enzyme deficiencies in the liver. The degree of hyperoxaluria is marked, and stone formation typically begins in childhood. Primary Hyperoxaluria (PH1) is a caused by deficiency of alanine glyoxylate aminotransferase (AGT), a hepatic enzyme that converts glyoxylate to glycine. Absence of AGT activity results in conversion of glyoxylate to oxalate, which is not capable of being degraded. Excess oxalate is excreted in the urine, causing kidney stones, nephrocalcinosis and kidney failure. As kidney function declines, blood levels of oxalate increase markedly and oxalate combines with calcium to form calcium oxalate deposits in the kidneys, eyes, heart, bones, and other organs, resulting in systemic disease. Primary hyperoxaluria type 2 (PH2) is caused by deficiency of the hepatic enzyme glyoxylate reductase/hydroxypyruvate reductase (GRHPR). Absence of GRHPR activity results in excess oxalate and usually L-glycerate excreted in the urine leading to kidney stones and sometimes kidney failure. It is less common than PH1. Onset of PH2 is typically in childhood or adolescence with
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
symptoms related to kidney stones. Patients with PH2 appear to have less
active stone formation and better preservation of kidney function when
compared to patients with PH1. End-stage kidney disease is also less common
and of later onset than in PH1 but once it happens oxalate deposition in other
organs such as bone, retina and myocardium can occur.
Early diagnosis of oxalosis is of immense value because at a stage when renal
failure has not set in, a proper management can arrest or at least delay the
progress of disease. Liver function tests may be abnormal in advanced
disease. Liver biopsy to demonstrate reduced enzyme activity is the
confirmatory test. Molecular diagnosis, being non-invasive, is preferred if
available. Long-term survival in patients is possible only with combined renal
and hepatic transplantation. In patients who have already developed renal
failure at the time of diagnosis of oxalosis, a combined liver and kidney
transplantation offers the most effective treatment because the new liver will
have the capability of producing necessary enzymes and the new kidney will
excrete oxalates normally. Dialysis alone as a modality of treatment has been
observed to be ineffective in retarding the disease progress because the
amount of oxalate production almost always surpasses the amount removable
by dialysis, thereby leading to a positive balance in favour of oxalate
deposition.
References:
Hoppe B. An update on Primary hyperoxaluria. Nat Rev Nephrol.2012;8(8):467-75
Hoppe B, Beck BB, Mil iner DS. The Primary hyperoxalurias. Kidney Int.2009;75(12); 1264-71.
Latta K, Brodeh L J : Primary hyperoxaluria type 1. Review Eu J Pediatr .1990; 149:518-522.
Lieske JC, Monico CG, Holmes WS, et al: International Registry for primary hyperoxaluria . Am J Nephrol 2005.May- Jun; 25(3): 290-296
Hoppe B , Langman CB : A united States survey on diagnosis, treatment and outcome of primary hyperoxaluria. Pediatr Nephrol 2003; 18: 986-991.
Alkhunaizi A and Chan L. Secondary oxalosis : a cause of delayed recovery of renal function in setting of acute renal failure . J Am Soc Nephrol . 1996 ; 7: 2321-2326.
Mashours , Turner JF Jr and Merrell R . Acute Renal failure, Oxalosis and Vitamin C supplementation . Chest 2000; 118 : 561-563.
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
Danpure CJ, Jennings PR: Peroxisomal alanine:glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Lett 1986;201: 20-24
Morgan SH, Purkiss P, Watts RWE, Mansell MA: Oxalate dynamics in chronic kidney failure. Comparison with normal subjects and patients with primary hyperoxaluria. Nephron 1987;46:253-257.
10. Hoppe B, Kemper MJ, Bokenkamp A, Langman CB: Plasma calcium-oxalate
saturation in children with renal insufficiency and in children with primary hyperoxaluria. Kidney Int 1998;54:921-925
11. Milliner DS, Wilson DM, Smith LH: Phenotypic expression of primary hyperoxaluria:
Comparative features of types I and II. Kidney Int 2001;59:31-36
12. Milliner DS, Eickholt JT, Bergstralh EJ, et al: Results of Long-Term Treatment with
Orthophosphate and Pyridoxine in Patients with Primary Hyperoxaluria. NEJM 1994;334:1553-1558
13. Wong PN, Tong GMW, Lo KY, Law ELK, Wong AKM. Primary hyperoxaluria: a rare
but important cause of nephrolihtiasis. HKMJ 2002; 8: 202-206.
14. Cochat P, Gaulier JM, Koch PC et al. Combined liver kidney transplantation in primary
hyperoxaluria type1. Eur J Pediatr 1999; 158[suppl]: 575-580.
15. Bunchman TE and Swartz RD. Oxalate removal in type 1 hyperoxaluria or acquired
oxalosis using HD and equilibration PD. Perit Dial Int. 1994; 14: 81-84.
16. Halil O, Farringdon K. Oxalosis: an unusual cause of leucoerythroblastic anemia. Br J
Haematol 2003;12
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
ATYPICAL CHRONIC MYELOID LEUKEMIA WITH t(9;22)
(P24,11.2), AND JAK2 GENE NEGATIVE
Tariq Mahmood Satti, Parvez Ahmed, Qamar un Nisa Chaudhry, Syed Kamran Ahmed, Mehreen Ali Khan,
Raheel Iftikhar, Armed Forces Bone Marrow Transplant Centre, Rawalpindi – Pakistan
Abstract
We report here on a rare case of BCR-ABL1 and t(9;22) negative atypical
chronic myeloid leukemia with sensitive to the tyrosine kinase inhibitors
imatinib mesylate. At two years of follow-up, the patient showed complete
hematologic response. Now at present his visceromegaly is reversed and
bone marrow is in complete haematological remission.
Introduction
Chronic Myeloid Leukemia (CML) is the best described disease resulting from the t(9;22)(q34,q11.2). This chromosomal rearrangement leads to the well-known BCR-ABL fusion that promotes tyrosine kinase activity. Other oncogenic BCR fusions have also been found, such as platelet-derived growth factor receptor-alpha gene (PDGFRA) (4q12)and fibroblast growth factor receptor 1 (FGFR1) (8p12), which cause myeloproliferative disorders (MD). The janus kinase 2 (JAK2) gene is one of the four members of the JAK gene family. The JAK2 V617F mutation which results from a G->T transversion at nucleotide 1849 in exon 14 of the JAK2 gene resulting in the substitution of valine by phenylalanine at codon 617, is associated with MD and is a major diagnostic criterion for primary myelofibrosis, polycythemia vera and essential thrombocythemia. A great number of chromosomal translocations involving the JAK2 locus have been described. We report here on an extremely rare case of atypical CML that was found to be breakpoint cluster region (BCR)-Abelson (ABL) 1 and JAK-2 negative. This is a quite rare case reported in literature.
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
Case Report
In April 2013, a 28-year-old male patient presented with fatigue, abdominal pain and splenomegaly. A blood count revealed leukocytosis (90.38 x 109/L) with a predominance of neutrophils and a shift to the left. Hypercellular bone marrow with granulocytic and erythroid dysplasia was described. Conventional cytogenetic analysis was performed and a 46,XY karyotype was found 1) In view of the clinical picture, the result was interpreted as indicative of the presence of a BCR-ABL1 fusion gene, but this was not detected by reverse transcription polymerase chain reaction (RT-PCR). The presence of a BCR-ABL rearrangement was also ruled out by fluorescence in situ hybridization (FISH) using a BCR-ABL probe. In addition, no BCR/PDGFR FIP fusion gene or JAK2 V617F mutation were detected by RT-PCR. Therefore, the case was classified, according to the latest World Health Organization Classification, as BCR-ABL1-negative atypical CML.
Figure 1 Conventional cytogenetics showing 46,XY karyotype
The patient was treated with imatinib at a dose of 400 mg/day. After three months of treatment, he presented with regression in spleen size and complete hematologic response. His medication was continued in same dose.
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
In December 2013, his bone marrow biopsy was done and it showed
hematologic remission, the imatinib treatment was continued.
Conventional karyotyping was performed again and the 46,XY, karyotype was
observed in all metaphases examined.
In December 2015, the patient had complete hematologic remission with
imatinib and he was advised to continue treatment.
Discussion
The patient described here, presenting BCR-ABL1 and t(9;22) negative
atypical CML was treated with the tyrosine kinase inhibitors imatinib but
achieved complete hematologic response. Of the ten previously reported
cases, five developed MF, two transformed into acute myeloid leukemia and
three remained unclassified MPN. Only one of them was treated with imatinib,
and the patient could not be followed-up. It is important to observe that in the
remaining cases the mortality rate was 70%. The factors includes promote
tumorigenic properties and lead to increased cell survival in this disorder is still
mystery.
It was not possible to define which therapy would be best because tyrosine
kinase inhibitors may not be effective in all cases. Some authors suggest that
in these cases it is important to investigate the role of interferon alone or in
combination with TKI. We decided to submit our patient to TKI (imatinib) alone
because he had no matched sibling donor.
Our case shows a rare t(9;22 and BCR/ABL negative patient with MD. This
finding is unable to explain the tumorigenic activity seen in preclinical studies.
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interest
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
References
Baxter EJ, Hochhaus A, Bolufer P, Reiter A, Fernandez JM, Senent L, et al. The t(4;22)(q12;q11) in atypical chronic myeloid leukaemia fuses BCRto PDGFRA. Hum Mol Genet. 2002;11(12):1391–1397.
Demiroglu A, Steer EJ, Heath C, Taylor K, Bentley M, Allen SL, et al. The t(8;22) in chronic myeloid leukemia fuses BCR to FGFR1: transforming activity and specific inhibition of FGFR1 fusion proteins.Blood. 2001;98(13):3778–3783.
Fioretos T, Panagopoulos I, Lassen C, Swedin A, Billström R, Isaksson M, et al. Fusion of the BCR and the fibroblast growth factor receptor-1 (FGFR1) genes as a result of t(8;22)(p11;q11) in a myeloproliferative disorder: The first fusion gene involving BCR but not ABL. Genes Chromosomes Cancer. 2001;32(4):302–310.
Thiele J, Kvasnicka HM, Orazi A, Tefferi A, Birgegard G. World Health Organization classification of tumours of haematopoietic and Lymphoid Tissues. Chronic myeloproliferative disorders treatment. 4. Lyon: International Agency for Research on Cancer Press; 2008.
Griesinger F, Hennig H, Hillmer F, Podleschny M, Steffens R, Pies A, et al. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11.2) translocation in a patient with a clinically typical chronic myeloid leukemia.Genes Chromosomes Cancer. 2005;44(3):329–333.
Cirmena G, Aliano S, Fugazza G, Bruzzone R, Garuti A, Bocciardi R, et al. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11) in a patient with acute myeloid leukemia. Cancer Genet Cytogenet.2008;183(2):105–108.
Lane SW, Fairbairn DJ, McCarthy C, Nandini A, Perry-Keene J, Kennedy GA. Leukaemia cutis in atypical chronic myeloid leukaemia with a t(9;22) (p24;q11.2) leading to BCR-JAK2 fusion. Br J Haematol.2008;142(4):503.
Elnaggar MM, Agersborg S, Sahoo T, Girgin A, Ma W, Rakkhit R, et al. BCR- JAK2 fusion as a result of a translocation (9;22)(p24;q11.2) in a patient with CML-like myeloproliferative disease. Mol Cytogenet.2012;5(1):23.
Tirado CA, Chen W, Huang LJ, Laborde C, Hiemenz MC, Valdez F, et al. Novel JAK2 rearrangement resulting from a t(9;22)(p24;q11.2) in B-acute lymphoblastic leukemia. Leuk Res. 2010;34(12):1674–1676.
10. Cuesta-Domínguez Á, Ortega M, Ormazábal C, Santos-Roncero M, Galán-Díez M,
Steegmann JL, et al. Transforming and tumorigenic activity of JAK2 by fusion to BCR: molecular mechanisms of action of a novel BCR-JAK2 tyrosine-kinase. PLoS One. 2012;7(2):e32451.
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
ACADEMIC ACTIVITIES AND EVENTS OF PSH FORUM:
Keeping alive the tradition of annual PSH conference, 17th PSH Conference
was held at Pearl Continental, Rawalpindi in March 2015. The conference was
organised by Armed Forces Institute of Pathology (Haematology Department).
The conference was well organised and was complete success. The main
anchor person of this organisation was Maj Gen Saleem Ahmed Khan and the
mentor was Maj Gen Mohammad Ayyub, HI(M). The some glimpses of the
conference are shown below.
17th Annual Conference Pakistan Society of Haematology (PSH)
17th Annual Conference Pakistan Society of Haematology (PSH)
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
PSH Newsletter
Volume: 10, No.1 Jan – Mar 2016
Dear Colleagues
We request you to join us in newsletter by sending your comments, short
communications, case reports, issues of national interest, new
developments in your departments, and scientific activities in your institutes.
Your contribution is the back bone of this newsletter. It is requested that
report/write up should be brief and concise. For information, suggestions,
and correspondences please e-mail to [email protected]. In
case you are a member and you are not receiving mails from us please
update your postal and e-mail address urgently. You can find us at
Website: www.psh.org.pk and Facebook: /psh.org.pk.
Brig (Dr) Tariq Mahmood Satti
Secretary
Pakistan Society of Haematology (PSH)
PAKISTAN SOCIETY OF HAEMATOLOGY – NEWSLETTER 2016
Source: http://psh.org.pk/forumfiles/Newsletter_2016.pdf
Peter F. Drucker defines innovation as "change that creates a "messy rather than Innovation is change that new dimension of performance." No matter what organizations orderly," and innova- creates a new dimension do, whether they are in business, government or the social sec- tion requires "courage of performance. tor, they must continuously develop new ideas and initiate
Journal of Controlled Release 94 (2004) 323 – 335 Incorporation and release behavior of hydrophobic drug in functionalized poly(D,L-lactide)-block–poly(ethylene oxide) Jaeyoung Lee, Eun Chul Cho, Kilwon Cho* Department of Chemical Engineering, School of Environmental Engineering, Pohang University of Science and Technology, 790-784 Pohang, South Korea Received 22 May 2003; accepted 9 October 2003

